
The ATP1A2 Mutation Associated with Hemiplegic Migraines: Case Report and Literature Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
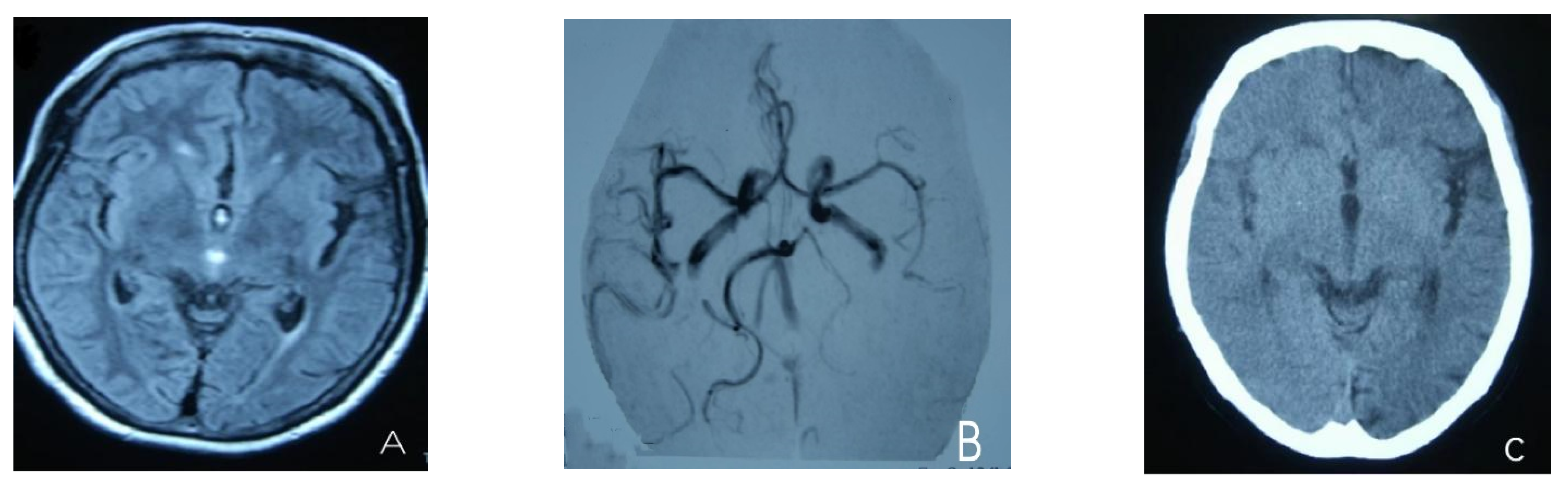
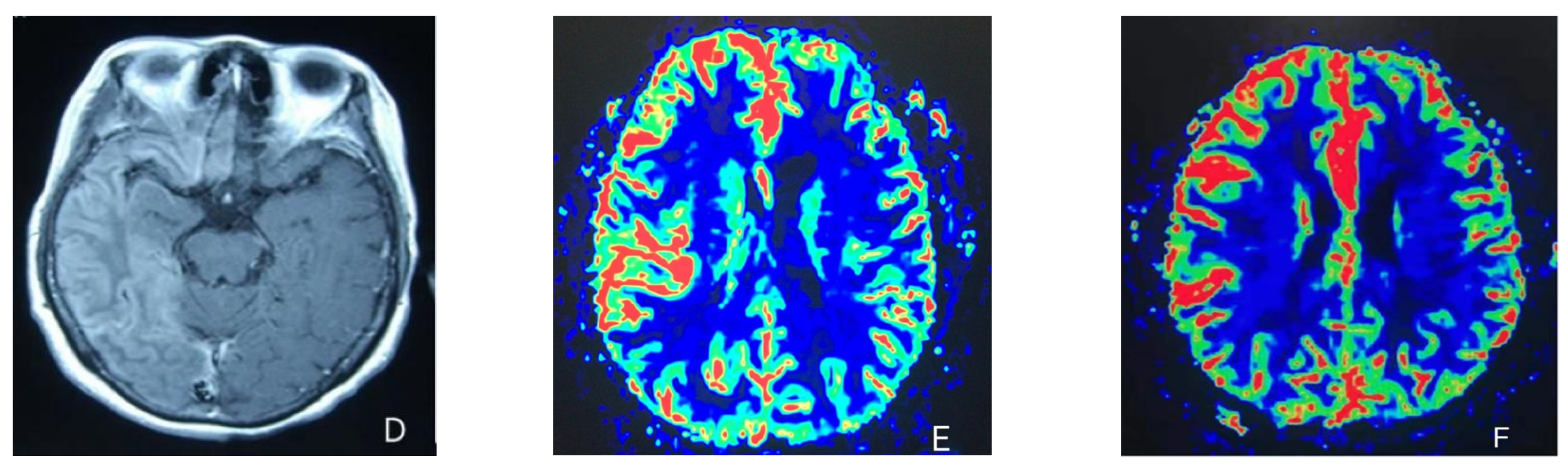
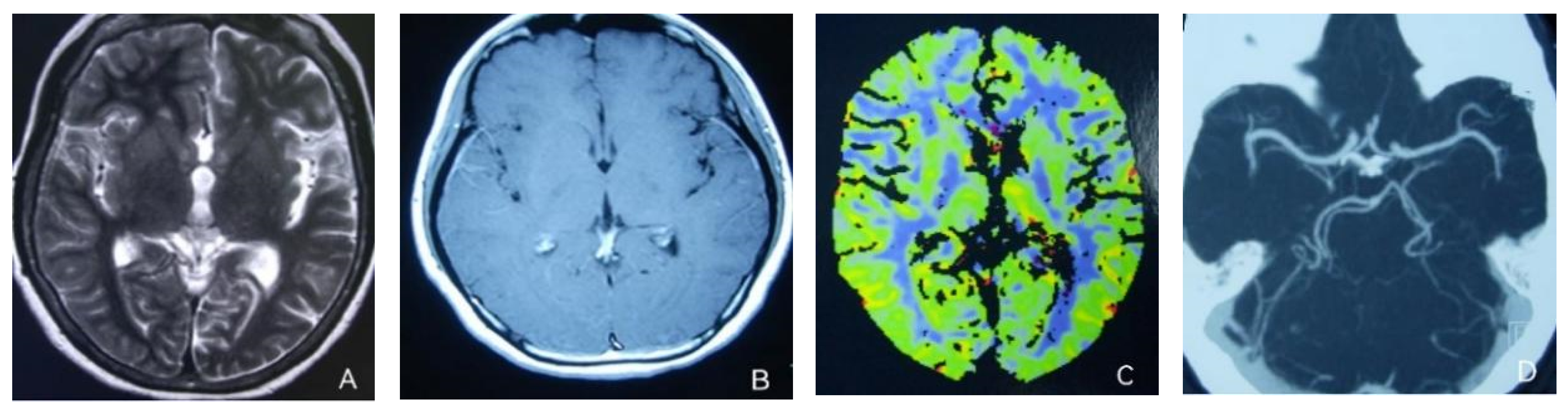
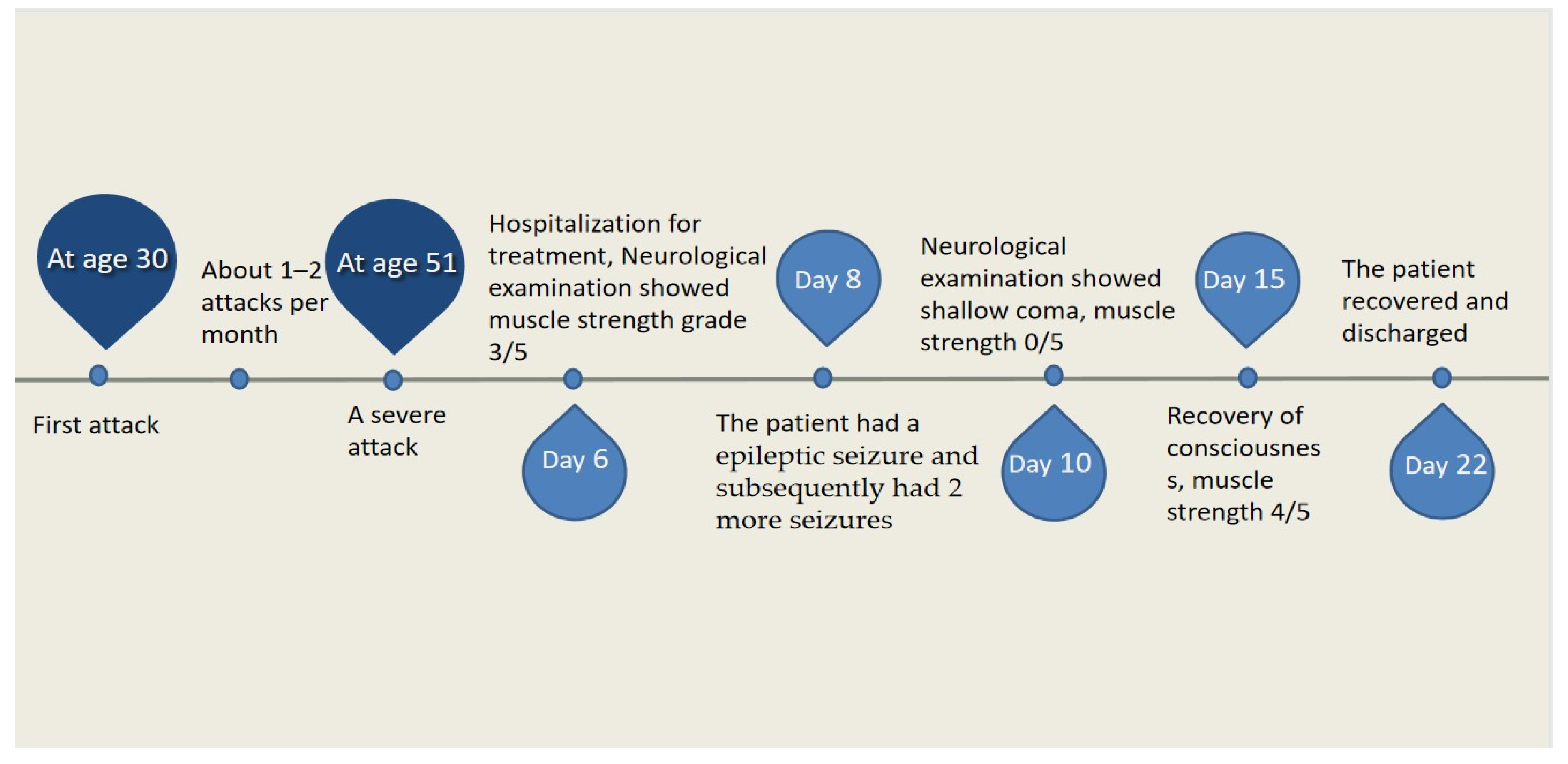
2. Case Presentation
3. Discussion
3.1. CSD and Aura
3.2. CSD and Migraine
3.3. CSD, ATP1A2 and Neurovascular Coupling
3.4. CSD, Seizure and Vasogenic Edema
3.5. CSD, ATP1A2 and Seizure
3.6. ATP1A2, CSD and Transient Motor Impairment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Stefano, V.; Rispoli, M.G.; Pellegrino, N.; Graziosi, A.; Rotondo, E.; Napoli, C.; Pietrobon, D.; Brighina, F.; Parisi, P. Diagnostic and therapeutic aspects of hemiplegic migraine. J. Neurol. Neurosurg. Psychiatry 2020, 91, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C. Cortical Spreading Depression Triggers Migraine Attack: Pro. Headache J. Head Face Pain 2010, 50, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Tavraz, N.N.; Junghans, C. ATP1A2 Mutations in Migraine: Seeing through the Facets of an Ion Pump onto the Neurobiology of Disease. Front. Physiol. 2016, 7, 239. [Google Scholar] [CrossRef]
- Unekawa, M.; Ikeda, K.; Tomita, Y.; Kawakami, K.; Suzuki, N. Enhanced susceptibility to cortical spreading depression in two types of Na+, K+-ATPase α2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 2018, 38, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Bertolero, M.A.; Liu, W.B.; D’Esposito, M. The Human Thalamus Is an Integrative Hub for Functional Brain Networks. J. Neurosci. 2017, 37, 5594–5607. [Google Scholar] [CrossRef]
- Sepulcre, J.; Sabuncu, M.R.; Yeo, T.B.; Liu, H.; Johnson, K.A. Stepwise Connectivity of the Modal Cortex Reveals the Multimodal Organization of the Human Brain. J. Neurosci. 2012, 32, 10649–10661. [Google Scholar] [CrossRef]
- Vuralli, D.; Karatas, H.; Yemisci, M.; Bolay, H. Updated review on the link between cortical spreading depression and headache disorders. Expert Rev. Neurother. 2021, 21, 1069–1084. [Google Scholar] [CrossRef]
- Silvestro, M.; Tessitore, A.; Di Nardo, F.; di Clemente, F.S.; Trojsi, F.; Cirillo, M.; Esposito, F.; Tedeschi, G.; Russo, A. Functional connectivity changes in complex migraine aura: Beyond the visual network. Eur. J. Neurol. 2022, 29, 295–304. [Google Scholar] [CrossRef]
- Levy, D.; Burstein, R.; Kainz, V.; Jakubowski, M.; Strassman, A.M. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 2007, 130, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Levy, D. Endogenous Mechanisms Underlying the Activation and Sensitization of Meningeal Nociceptors: The Role of Immuno-Vascular Interactions and Cortical Spreading Depression. Curr. Pain Headache Rep. 2012, 16, 270–277. [Google Scholar] [CrossRef]
- Waeber, C.; Moskowitz, M.A. Migraine as an inflammatory disorder. Neurology 2005, 64, S9–S15. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Strassman, A.M.; Burstein, R.; Levy, D. Sensitization and Activation of Intracranial Meningeal Nociceptors by Mast Cell Mediators. J. Pharmacol. Exp. Ther. 2007, 322, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Erdener, Ş.E.; Kaya, Z.; Dalkara, T. Parenchymal neuroinflammatory signaling and dural neurogenic inflammation in migraine. J. Headache Pain 2021, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Tominaga, N.; Kaneko, J.; Sato, M.; Akutsu, T.; Hamada, J.; Sakai, F.; Nishiyama, K. Biphasic neurovascular changes in prolonged migraine aura in familial hemiplegic migraine type 2. J. Neurol. Neurosurg. Psychiatry 2015, 86, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Colinas, O.; Moreno-Domínguez, A.; Zhu, H.-L.; Walsh, E.J.; Pérez-García, M.T.; Walsh, M.P.; Cole, W.C. α5-Integrin-mediated cellular signaling contributes to the myogenic response of cerebral resistance arteries. Biochem. Pharmacol. 2015, 97, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Lauritzen, M. Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Lai, F.; Banerjee, M.; Duan, Q.; Li, Z.; Si, S.; Xie, Z. Expression of Mutant α1 Na/K-ATPase Defective in Conformational Transition Attenuates Src-mediated Signal Transduction*. J. Biol. Chem. 2013, 288, 5803–5814. [Google Scholar] [CrossRef]
- Staehr, C.; Hangaard, L.; Bouzinova, E.V.; Kim, S.; Rajanathan, R.; Jessen, P.B.; Luque, N.; Xie, Z.; Lykke-Hartmann, K.; Sandow, S.L.; et al. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2019, 39, 1570–1587. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef]
- Longden, T.A.; Nelson, M.T. Vascular Inward Rectifier K+ Channels as External K+ Sensors in the Control of Cerebral Blood Flow. Microcirculation 2015, 22, 183–196. [Google Scholar] [CrossRef]
- Hosford, P.S.; Gourine, A.V. What is the key mediator of the neurovascular coupling response? Neurosci. Biobehav. Rev. 2019, 96, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Staehr, C.; Rajanathan, R.; Postnov, D.D.; Hangaard, L.; Bouzinova, E.V.; Lykke-Hartmann, K.; Bach, F.W.; Sandow, S.L.; Aalkjaer, C.; Matchkov, V.V. Abnormal neurovascular coupling as a cause of excess cerebral vasodilation in familial migraine. Cardiovasc. Res. 2020, 116, 2009–2020. [Google Scholar] [CrossRef]
- Roth, C.; Ferbert, A.; Huegens-Penzel, M.; Siekmann, R.; Freilinger, T. Multimodal imaging findings during severe attacks of familial hemiplegic migraine type 2. J. Neurol. Sci. 2018, 392, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gürsoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Soldner, E.L.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef]
- Seiffert, E. Lasting Blood-Brain Barrier Disruption Induces Epileptic Focus in the Rat Somatosensory Cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef]
- Broekaart, D.W.; Bertran, A.; Jia, S.; Korotkov, A.; Senkov, O.; Bongaarts, A.; Mills, J.D.; Anink, J.J.; Seco-Moral, J.; Baayen, J.C.; et al. The matrix metalloproteinase inhibitor IPR-179 has antiseizure and antiepileptogenic effects. J. Clin. Investig. 2021, 131, e138332. [Google Scholar] [CrossRef]
- Pisano, T.; Spiller, S.; Mei, D.; Guerrini, R.; Cianchetti, C.; Friedrich, T.; Pruna, D. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia 2013, 33, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Kros, L.; Lykke-Hartmann, K.; Khodakhah, K. Increased susceptibility to cortical spreading depression and epileptiform activity in a mouse model for FHM2. Sci. Rep. 2018, 8, 16959. [Google Scholar] [CrossRef]
- Wei, Y.; Ullah, G.; Schiff, S.J. Unification of Neuronal Spikes, Seizures, and Spreading Depression. J. Neurosci. 2014, 34, 11733–11743. [Google Scholar] [CrossRef] [PubMed]
- Ullah, G.; Wei, Y.; Dahlem, M.A.; Wechselberger, M.; Schiff, S.J. The Role of Cell Volume in the Dynamics of Seizure, Spreading Depression, and Anoxic Depolarization. PLoS Comput. Biol. 2015, 11, e1004414. [Google Scholar] [CrossRef]
- Tamim, I.; Chung, D.Y.; de Morais, A.L.; Loonen, I.C.M.; Qin, T.; Misra, A.; Schlunk, F.; Endres, M.; Schiff, S.J.; Ayata, C. Spreading depression as an innate antiseizure mechanism. Nat. Commun. 2021, 12, 2206. [Google Scholar] [CrossRef]
- Hasırcı Bayır, B.R.; Tutkavul, K.; Eser, M.; Baykan, B. Epilepsy in patients with familial hemiplegic migraine. Seizure 2021, 88, 87–94. [Google Scholar] [CrossRef]
- Mantegazza, M.; Cestèle, S. Pathophysiological mechanisms of migraine and epilepsy: Similarities and differences. Neurosci. Lett. 2018, 667, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Loonen, I.; Baca, S.M.; Schenke, M.; Meijer, J.H.; Ferrari, M.D.; Terwindt, G.M.; Voskuyl, R.A.; Charles, A.; Maagdenberg, A.M.V.D.; et al. Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J. Cereb. Blood Flow Metab. 2017, 37, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Chen, X.; Brier, L.M.; Bumstead, J.R.; Rensing, N.R.; Ringel, A.E.; Shin, H.; Oldenborg, A.; Crowley, J.R.; Bice, A.R.; et al. Astrocyte deletion of α2-Na/K ATPase triggers episodic motor paralysis in mice via a metabolic pathway. Nat. Commun. 2020, 11, 6164. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.-P.; Oliet, S.H. Synaptic and Extrasynaptic NMDA Receptors Are Gated by Different Endogenous Coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Yue, W. The ATP1A2 Mutation Associated with Hemiplegic Migraines: Case Report and Literature Review. Clin. Transl. Neurosci. 2022, 6, 25. https://doi.org/10.3390/ctn6040025
Liu C, Yue W. The ATP1A2 Mutation Associated with Hemiplegic Migraines: Case Report and Literature Review. Clinical and Translational Neuroscience. 2022; 6(4):25. https://doi.org/10.3390/ctn6040025
Chicago/Turabian StyleLiu, Changyue, and Wei Yue. 2022. "The ATP1A2 Mutation Associated with Hemiplegic Migraines: Case Report and Literature Review" Clinical and Translational Neuroscience 6, no. 4: 25. https://doi.org/10.3390/ctn6040025
APA StyleLiu, C., & Yue, W. (2022). The ATP1A2 Mutation Associated with Hemiplegic Migraines: Case Report and Literature Review. Clinical and Translational Neuroscience, 6(4), 25. https://doi.org/10.3390/ctn6040025