Cluster Headache Pathophysiology—A Disorder of Network Excitability?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
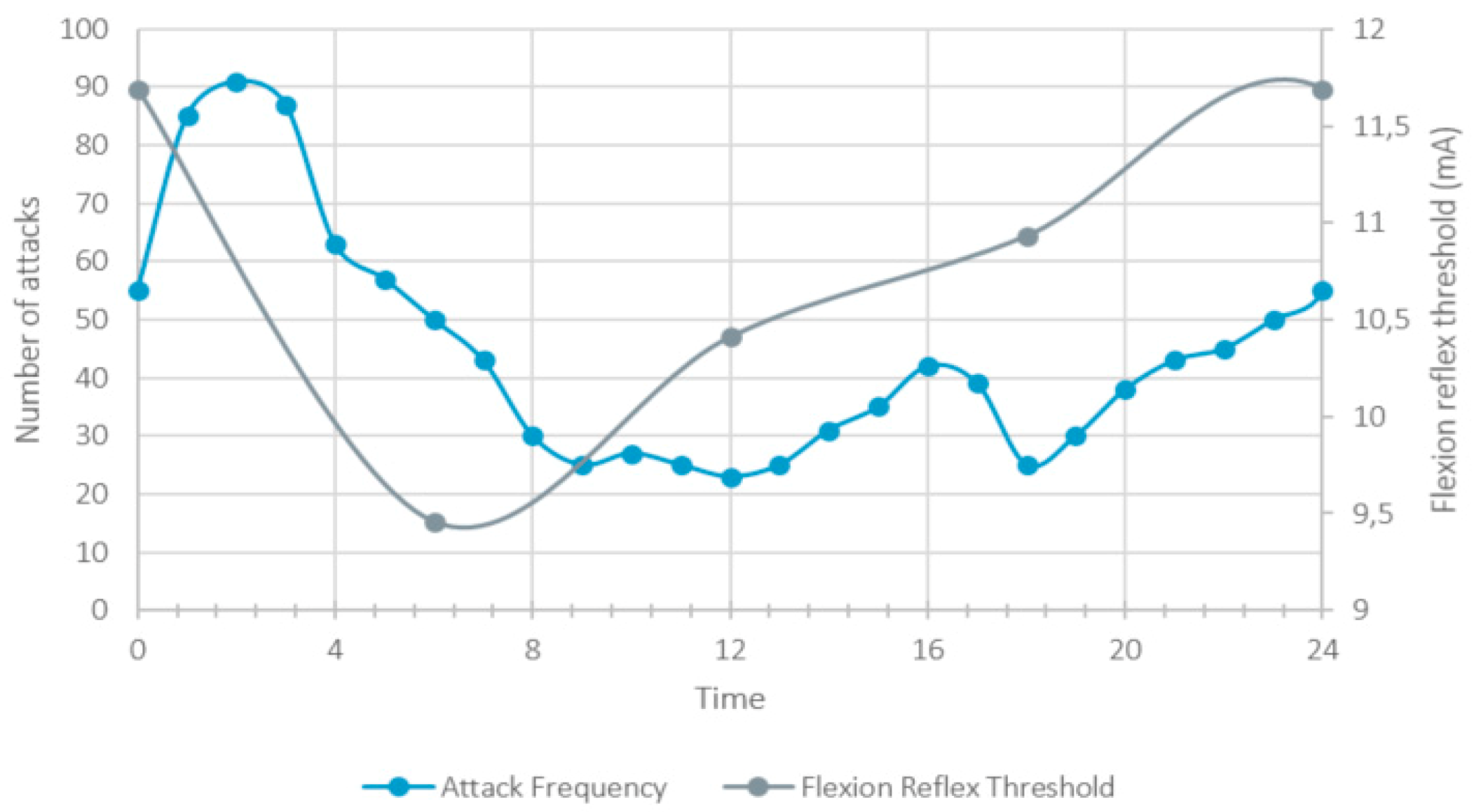
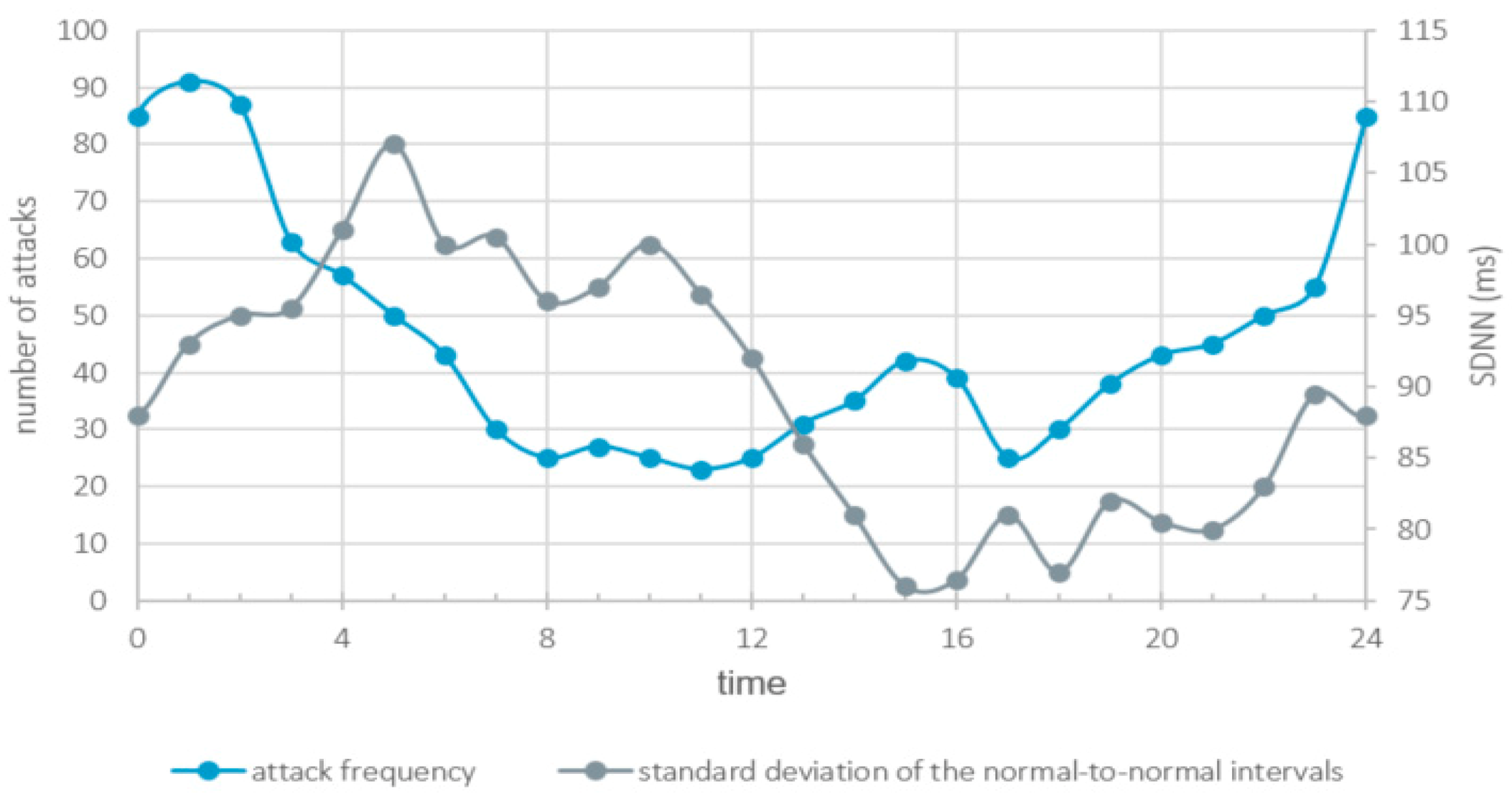
2. Pain and Restlessness
3. Sensitization
4. The Influence of Prophylactic Treatment
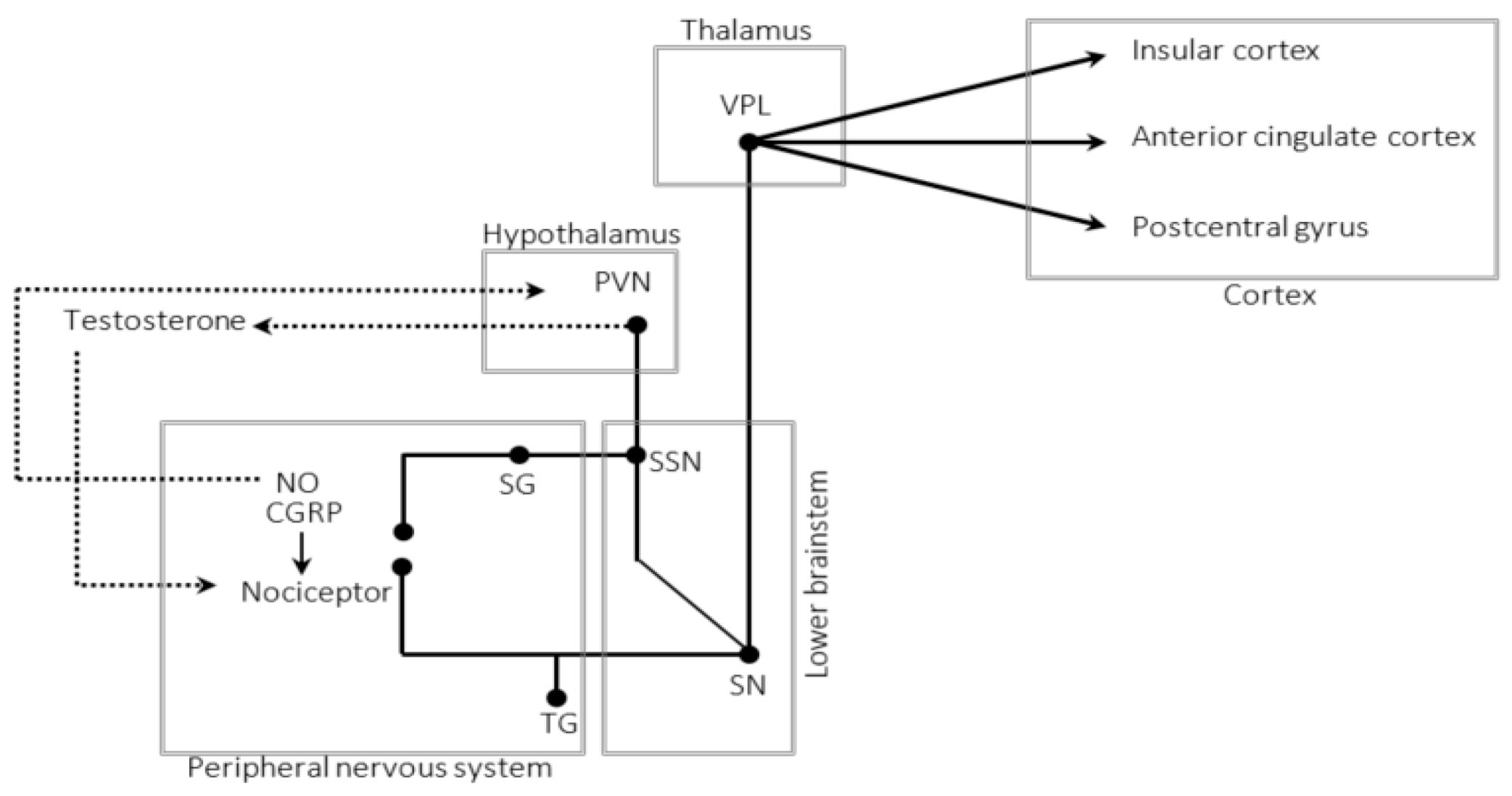
5. The Trigeminal–Autonomic Reflex
6. Circadian Rhythmicity
7. The Autonomic Nervous System
8. The Hypothalamus
9. Homeostatic Plasticity and the Occurrence of Attacks
10. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CGRP | calcitonin gene-related peptide |
| CH | Cluster headache |
| CSF | cerebrospinal fluid |
| fMRI | functional magnetic resonance imaging |
| GABA | gamma-aminobutyric acid |
| NFR | nociceptive flexion reflex |
| NO | nitric oxide |
| PAG | periaqueductal grey |
| PET | positron emission tomography |
| PVN | paraventricular nucleus |
| QST | quantitative sensory testing |
| SDNN | standard deviation of the normal-to-normal intervals |
| STD | short-term depression |
| VGCC | voltage-gated calcium channels |
| VIP | vasoactive intestinal polypeptide |
References
- Schröder, P.; Gaul, C.; Drabik, A.; Molsberger, A. Pain localization in cluster headache patients: Onset, peak, and radiation. Acta Neurol. Scand. 2021, 143, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Burish, M.J.; Pearson, S.M.; Shapiro, R.E.; Zhang, W.; Schor, L.I. Cluster headache is one of the most intensely painful human conditions: Results from the International Cluster Headache Questionnaire. Headache J. Head Face Pain 2021, 61, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J. Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar]
- Pedersen, A.S.; Snoer, A.; Barloese, M.; Petersen, A.; Jensen, R.H. Prevalence of pre-cluster symptoms in episodic cluster headache: Is it possible to predict an upcoming bout? Cephalalgia 2021, 41, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Barloese, M.; Lund, N.; Petersen, A.; Rasmussen, M.; Jennum, P.; Jensen, R. Sleep and chronobiology in cluster headache. Cephalalgia 2015, 35, 969–978. [Google Scholar] [CrossRef]
- Lund, N.; Barloese, M.; Petersen, A.; Haddock, B.; Jensen, R. Chronobiology differs between men and women with cluster headache, clinical phenotype does not. Neurology 2017, 88, 1069–1076. [Google Scholar] [CrossRef]
- Lee, M.J.; Choi, H.A.; Shin, J.H.; Park, H.R.; Chung, C.-S. Natural course of untreated cluster headache: A retrospective cohort study. Cephalalgia 2017, 38, 655–661. [Google Scholar] [CrossRef]
- Morelli, N.; Rota, E.; Gori, S.; Guidetti, D.; Michieletti, E.; DE Simone, R.; Di Salle, F. Brainstem activation in cluster headache: An adaptive behavioural response? Cephalalgia 2013, 33, 416–420. [Google Scholar] [CrossRef]
- May, A.; Bahra, A.; Büchel, C.; Frackowiak, R.; Goadsby, P.J. Hypothalamic activation in cluster headache attacks. Lancet 1998, 352, 275–278. [Google Scholar] [CrossRef]
- Legrain, V.; Iannetti, G.D.; Plaghki, L.; Mouraux, A. The pain matrix reloaded: A salience detection system for the body. Prog. Neurobiol. 2011, 93, 111–124. [Google Scholar] [CrossRef]
- Frederickson, R.C.; Burgis, V.; Edwards, J.D. Hyperalgesia induced by naloxone follows diurnal rhythm in responsivity to painful stimuli. Science 1977, 198, 756–758. [Google Scholar] [CrossRef]
- Göbel, H.; Cordes, P.; Christiani, K. Algesimetric experiment on pain sensitivity in the pericranial musculature. Circadian rhythms and corresponding psychic variables. Schmerz 1989, 3, 209–218. [Google Scholar] [CrossRef]
- Göbel, H.; Cordes, P. Circadian Variation of Pain Sensitivity in Pericranial Musculature. Headache J. Head Face Pain 1990, 30, 418–422. [Google Scholar] [CrossRef]
- Sandrini, G.; Alfonsi, E.; Bono, G.; Facchinetti, F.; Montalbetti, L.; Nappi, G. Circadian variations of human flexion reflex. Pain 1986, 25, 403–410. [Google Scholar] [CrossRef]
- Aviram, J.; Shochat, T.; Pud, D. Pain Perception in Healthy Young Men Is Modified by Time-Of-Day and Is Modality Dependent. Pain Med. 2015, 16, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, G.; Vanier, M.-C. Biological rhythms in pain and in the effects of opioid analgesics. Pharmacol. Ther. 1995, 68, 129–147. [Google Scholar] [CrossRef]
- Bruguerolle, B.; Labrecque, G. Rhythmic pattern in pain and their chronotherapy. Adv. Drug Deliv. Rev. 2007, 59, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Rolke, R.; Baron, R.; Maier, C.; Tölle, T.R.; Treede, R.-D.; Beyer, A.; Binder, A.; Birbaumer, N.; Birklein, F.; Bötefür, I.C.; et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): Standardized protocol and reference values. Pain 2006, 123, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, A.; Coppola, G.; Anastasio, M.G.; De Icco, R.; Ambrosini, A.; Serrao, M.; Parisi, V.; Evangelista, M.; Sandrini, G.; Pierelli, F.; et al. Trait- and Frequency-Dependent Dysfunctional Habituation to Trigeminal Nociceptive Stimulation in Trigeminal Autonomic Cephalalgias. J. Pain 2018, 19, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Di Lorenzo, C.; Bracaglia, M.; Di Lenola, D.; Parisi, V.; Perrotta, A.; Serrao, M.; Pierelli, F. Lateralized nociceptive blink reflex habituation deficit in episodic cluster headache: Correlations with clinical features. Cephalalgia 2014, 35, 600–607. [Google Scholar] [CrossRef]
- Holle, D.; Zillessen, S.; Gaul, C.; Naegel, S.; Kaube, H.; Diener, H.-C.; Katsarava, Z.; Obermann, M. Habituation of the nociceptive blink reflex in episodic and chronic cluster headache. Cephalalgia 2012, 32, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Montagna, P.; Pierangeli, G.; Cortelli, P. The Primary Headaches as a Reflection of Genetic Darwinian Adaptive Behavioral Responses. Headache J. Head Face Pain 2010, 50, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Beissner, F.; Brandau, A.; Henke, C.; Felden, L.; Baumgartner, U.; Treede, R.D.; Oertel, B.G.; Lotsch, J. Quick discrimination of A(delta) and C fiber mediated pain based on three verbal descriptors. PLoS ONE 2010, 5, e12944. [Google Scholar] [CrossRef] [PubMed]
- Torelli, P.; Manzoni, G.C. Pain and behaviour in cluster headache. A prospective study and review of the literature. Funct. Neurol. 2004, 18, 205–210. [Google Scholar]
- Schurks, M.; Kurth, T.; De Jesus, J.; Jonjic, M.; Rosskopf, D.; Diener, H.-C. Cluster Headache: Clinical Presentation, Lifestyle Features, and Medical Treatment. Headache J. Head Face Pain 2006, 46, 1246–1254. [Google Scholar] [CrossRef]
- Blau, J. Behaviour during a cluster headache. Lancet 1993, 342, 723–725. [Google Scholar] [CrossRef]
- Keay, K.A.; Bandler, R. Distinct central representations of inescapable and escapable pain: Observations and speculation. Exp. Physiol. 2002, 87, 275–279. [Google Scholar] [CrossRef]
- Parry, D.; Macmillan, F.; Koutsikou, S.; McMullan, S.; Lumb, B. Separation of A- versus C-nociceptive inputs into spinal–brainstem circuits. Neuroscience 2008, 152, 1076–1085. [Google Scholar] [CrossRef]
- Bejjani, B.; Houeto, J.; Hariz, M.; Yelnik, J.; Mesnage, V.; Bonnet, A.; Pidoux, B.; Dormont, D.; Cornu, P.; Agid, Y. Aggressive behavior induced by intraoperative stimulation in the triangle of Sano. Neurology 2002, 59, 1425–1427. [Google Scholar] [CrossRef]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature 1983, 306, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Skljarevski, V.; Ramadan, N.M. The nociceptive flexion reflex in humans—Review article. Pain 2002, 96, 3–8. [Google Scholar] [CrossRef]
- Arendt-Nielsen, L.; Brennum, J.; Sindrup, S.; Bak, P. Electrophysiological and psychophysical quantification of temporal summation in the human nociceptive system. Graefes Arch. Clin. Exp. Ophthalmol. 1994, 68, 266–273. [Google Scholar] [CrossRef]
- Sandrini, G.; Antonaci, F.; Lanfranchi, S.; Milanov, I.; Danilov, A.; Nappi, G. Asymmetrical reduction of the nociceptive flexion reflex threshold in cluster headache. Cephalalgia 2000, 20, 647–652. [Google Scholar] [CrossRef]
- Nappi, G.; Sandrini, G.; Alfonsi, E.; Cecchini, A.P.; Micieli, G.; Moglia, A. Impaired Circadian Rhythmicity of Nociceptive Reflex Threshold in Cluster Headache. Headache J. Head Face Pain 2002, 42, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Sandrini, G.; Alfonsi, E.; Ruiz, L.; Pavesi, G.; Micieli, G.; Manzoni, G.C.; Mancia, D.; Nappi, G. Impairment of corneal pain perception in cluster headache. Pain 1991, 47, 299–304. [Google Scholar] [CrossRef]
- Ladda, J.; Straube, A.; Förderreuther, S.; Krause, P.; Eggert, T. Quantitative Sensory Testing in Cluster Headache: Increased Sensory Thresholds. Cephalalgia 2006, 26, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-T.; Donaldson, L.F.; Lumb, B.M. Differential contributions of A- and C-nociceptors to primary and secondary inflammatory hypersensitivity in the rat. Pain 2015, 156, 1074–1083. [Google Scholar] [CrossRef]
- Marmura, M.J.; Pello, S.J.; Young, W.B. Interictal pain in cluster headache. Cephalalgia 2010, 30, 1531–1534. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Young, W.B. Dynamic Mechanical (Brush) Allodynia in Cluster Headache. Headache J. Head Face Pain 2004, 44, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Göbel, C.H.; Karstedt, S.; Heinze, A.; Koch, B.; Göbel, H. Phenotype of Cluster Headache: Clinical Variability, Persisting Pain Between Attacks, and Comorbidities—An Observational Cohort Study in 825 Patients. Pain Ther. 2021, 1–17. [Google Scholar] [CrossRef]
- Ekbom, K. Abortive attacks in episodes of cluster headache. Arch. Neurobiol. 1974, 37, 191–195. [Google Scholar]
- Zamponi, G.W.; Lewis, R.J.; Todorovic, S.M.; Arneric, S.P.; Snutch, T.P. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 2009, 60, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Tamaddonfard, E.; Erfanparast, A.; Taati, M.; Dabbaghi, M. Role of opioid system in verapamil-induced antinociception in a rat model of orofacial pain. Veter. Res. Forum Int. Q. J. 2014, 5, 49–54. [Google Scholar]
- Vanegas, H.; Schaible, H.-G. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain 2000, 85, 9–18. [Google Scholar] [CrossRef]
- Hering, R.; Kuritzky, A. Sodium valproate in the treatment of cluster headache: An open clinical trial. Cephalalgia 1989, 9, 195–198. [Google Scholar] [CrossRef]
- Ximenes, J.C.M.; Gonçalves, D.D.O.; Siqueira, R.M.P.; Neves, K.R.T.; Cerqueira, G.S.; Correia, A.O.; Félix, F.H.C.; Leal, L.K.A.M.; Brito, G.A.D.C.; Naffah-Mazzacorati, M.D.G.; et al. Valproic acid: An anticonvulsant drug with potent antinociceptive and anti-inflammatory properties. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 575–587. [Google Scholar] [CrossRef]
- Kunder, S.K.; Bairy, L.K.; Arivazhahan, A. Effect of Sodium Valproate and Docosahexaenoic Acid on Pain in Rats. J. Clin. Diagn. Res. 2017, 11, FF05–FF08. [Google Scholar] [CrossRef]
- Hobo, S.; Eisenach, J.C.; Hayashida, K.-I. Up-regulation of spinal glutamate transporters contributes to anti-hypersensitive effects of valproate in rats after peripheral nerve injury. Neurosci. Lett. 2011, 502, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Stefani, L.C.; Muller, S.; Torres, I.L.; Razzolini, B.; Rozisky, J.R.; Fregni, F.; Markus, R.; Caumo, W. A Phase II, Randomized, Double-Blind, Placebo Controlled, Dose-Response Trial of the Melatonin Effect on the Pain Threshold of Healthy Subjects. PLoS ONE 2013, 8, e74107. [Google Scholar] [CrossRef]
- Miranda-Páez, A.; Zamudio, S.R.; Vázquez-León, P.; Sandoval-Herrera, V.; Villanueva-Becerril, I.; Carli, G. Effect of melatonin injection into the periaqueductal gray on antinociception and tonic immobility in male rats. Horm. Behav. 2017, 89, 23–29. [Google Scholar] [CrossRef]
- Leone, M.; D’Amico, D.; Moschiano, F.; Fraschini, F.; Bussone, G. Melatonin versus placebo in the prophylaxis of cluster headache: A double-blind pilot study with parallel groups. Cephalalgia 1996, 16, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Peres, M.; Rozen, T.D. Melatonin in the Preventive Treatment of Chronic Cluster Headache. Cephalalgia 2001, 21, 993–995. [Google Scholar] [CrossRef] [PubMed]
- Pringsheim, T.; Magnoux, E.; Dobson, C.F.; Hamel, E.; Aubé, M. Melatonin as adjunctive therapy in the prophylaxis of cluster headache: A pilot study. Headache J. Head Face Pain 2002, 42, 787–792. [Google Scholar] [CrossRef]
- Steiner, T.J.; Hering, R.; Couturier, E.; Davies, P.; Whitmarsh, T.E. Double-Blind Placebo-Controlled Trial of Lithium in Episodic Cluster Headache. Cephalalgia 1997, 17, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Stochino, M.E.; Deidda, A.; Asuni, C.; Cherchi, A.; Manchia, M.; Del Zompo, M. Evaluation of Lithium Response in Episodic Cluster Headache: A Retrospective Case Series. Headache J. Head Face Pain 2012, 52, 1171–1175. [Google Scholar] [CrossRef]
- Reilly, D. Lithium Vs Placebo in Cluster Headache. Cephalalgia 1998, 18, 1. [Google Scholar] [CrossRef]
- Weinsanto, I.; Mouheiche, J.; Laux-Biehlmann, A.; Aouad, M.; Maduna, T.; Petit-Demoulière, N.; Chavant, V.; Poisbeau, P.; Darbon, P.; Charlet, A.; et al. Lithium reverses mechanical allodynia through a mu opioid-dependent mechanism. Mol. Pain 2018, 14, 1744806917754142. [Google Scholar] [CrossRef]
- Banafshe, H.R.; Mesdaghinia, A.; Arani, M.N.; Ramezani, M.H.; Heydari, A.; Hamidi, G.A. Lithium attenuates pain-related behavior in a rat model of neuropathic pain: Possible involvement of opioid system. Pharmacol. Biochem. Behav. 2012, 100, 425–430. [Google Scholar] [CrossRef]
- Shimizu, T.; Shibata, M.; Wakisaka, S.; Inoue, T.; Mashimo, T.; Yoshiya, I. Intrathecal lithium reduces neuropathic pain responses in a rat model of peripheral neuropathy. Pain 2000, 85, 59–64. [Google Scholar] [CrossRef]
- Greco, R.; Mangione, A.; Siani, F.; Blandini, F.; Vairetti, M.; Nappi, G.; Sandrini, G.; Buzzi, M.; Tassorelli, C. Effects of CGRP receptor antagonism in nitroglycerin-induced hyperalgesia. Cephalalgia 2013, 34, 594–604. [Google Scholar] [CrossRef]
- Hebestreit, J.M.; May, A. Topiramate modulates trigeminal pain processing in thalamo-cortical networks in humans after single dose administration. PLoS ONE 2017, 12, e0184406. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Dodick, D.; Rigamonti, A.; D’Amico, D.; Grazzi, L.; Mea, E.; Bussone, G. Topiramate in Cluster Headache Prophylaxis: An Open Trial. Cephalalgia 2003, 23, 1001–1002. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.D.; Carrazana, E.J. Topiramate-treated cluster headache. Neurology 1999, 53, 234. [Google Scholar] [CrossRef] [PubMed]
- Förderreuther, S.; Mayer, M.; Straube, A. Treatment of Cluster Headache with Topiramate: Effects and Side-Effects in Five Patients. Cephalalgia 2002, 22, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Láinez, M.J.A.; Pascual, J.; Pascual, A.M.; Santonja, J.M.; Ponz, A.; Salvador, A. Topiramate in the Prophylactic Treatment of Cluster Headache. Headache J. Head Face Pain 2003, 43, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Lo, M.C.; Wang, S.J.; Tsai, J.J.; Wu, H.M. Topiramate in prevention of cluster headache in the Taiwanese. Neurol. India 2010, 58, 284–287. [Google Scholar] [CrossRef]
- Kuzniecky, R.; Hetherington, H.; Ho, S.; Pan, J.; Martin, R.; Gilliam, F.; Hugg, J.; Faught, E. Topiramate increases cerebral GABA in healthy humans. Neurology 1998, 51, 627–629. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L. Human in vivo evidence for trigeminovascular activation in cluster headache Neuropeptide changes and effects of acute attacks therapies. Brain 1994, 117, 427–434. [Google Scholar] [CrossRef]
- Goadsby, P.J. Cluster headache and the trigeminal-autonomic reflex: Driving or being driven? Cephalalgia 2017, 38, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.; Haji, A.A.; Hoffmann, J.; May, A. Peripheral provocation of cranial autonomic symptoms is not sufficient to trigger cluster headache attacks. Cephalalgia 2017, 38, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Matharu, M.; Goadsby, P.J. Persistence of attacks of cluster headache after trigeminal nerve root section. Brain 2002, 125, 976–984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jarrar, R.; Black, D.; Dodick, D.; Davis, D. Outcome of trigeminal nerve section in the treatment of chronic cluster headache. Neurology 2003, 60, 1360–1362. [Google Scholar] [CrossRef]
- Jürgens, T.P.; Barloese, M.; May, A.; Láinez, J.M.; Schoenen, J.; Gaul, C.; Goodman, A.M.; Caparso, A.; Jensen, R.H. Long-term effectiveness of sphenopalatine ganglion stimulation for cluster headache. Cephalalgia 2016, 37, 423–434. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Uddman, R.; Edvinsson, L. Cerebral vasodilatation in the cat involves nitric oxide from parasympathetic nerves. Brain Res. 1996, 707, 110–118. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Dodick, D.W.; Leone, M.; Bardos, J.N.; Oakes, T.M.; Millen, B.A.; Zhou, C.; Dowsett, S.A.; Aurora, S.K.; Ahn, A.H.; et al. Trial of Galcanezumab in Prevention of Episodic Cluster Headache. N. Engl. J. Med. 2019, 381, 132–141. [Google Scholar] [CrossRef]
- Teva Branded Pharmaceutical Products R&D, Inc. A Study to Explore the Long-Term Safety and Efficacy of Fremanezumab (TEV-48125) for the Prevention of Cluster Headache (ENFORCE). Available online: https://clinicaltrials.gov/ct2/show/NCT03107052 (accessed on 4 December 2020).
- Dodick, D.W.; Goadsby, P.J.; Lucas, C.; Jensen, R.; Bardos, J.N.; Martinez, J.M.; Zhou, C.; Aurora, S.K.; Yang, J.Y.; Conley, R.R.; et al. Phase 3 randomized, placebo-controlled study of galcanezumab in patients with chronic cluster headache: Results from 3-month double-blind treatment. Cephalalgia 2020, 40, 935–948. [Google Scholar] [CrossRef]
- Snoer, A.; Vollesen, A.L.H.; Beske, R.P.; Guo, S.; Hoffmann, J.; Fahrenkrug, J.; Jørgensen, N.R.; Martinussen, T.; Jensen, R.H.; Ashina, M. Calcitonin gene-related peptide and disease activity in cluster headache. Cephalalgia 2019, 39, 575–584. [Google Scholar] [CrossRef]
- Costa, A.; Ravaglia, S.; Sances, G.; Antonaci, F.; Pucci, E.; Nappi, G. Nitric Oxide Pathway and Response to Nitroglycerin in Cluster Headache Patients: Plasma Nitrite and Citrulline Levels. Cephalalgia 2003, 23, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, K.; Sjöstrand, C.; Svensson, D.A.; Waldenlind, E. Periods of Cluster Headache Induced by Nitrate Therapy and Spontaneous Remission of Angina Pectoris During Active Clusters. Cephalalgia 2004, 24, 92–98. [Google Scholar] [CrossRef]
- Steinberg, A.; Remahl, A.I.M.N. Role of Nitric Oxide in Cluster Headache. Curr. Pain Headache Rep. 2012, 16, 185–190. [Google Scholar] [CrossRef]
- Lin, Q.; Paleček, J.; Palečková, V.; Peng, Y.B.; Wu, J.; Cui, M.; Willis, W.D. Nitric oxide mediates the central sensitization of primate spinothalamic tract neurons. J. Neurophysiol. 1999, 81, 1075–1085. [Google Scholar] [CrossRef]
- Kitto, K.F.; Haley, J.E.; Wilcox, G.L. Involvement of nitric oxide in spinally mediated hyperalgesia in the mouse. Neurosci. Lett. 1992, 148, 1–5. [Google Scholar] [CrossRef]
- Meller, S.T.; Gebhart, G.F. Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain 1993, 52, 127–136. [Google Scholar] [CrossRef]
- Balgetir, F.; Avcı, D.; Gönen, M.; Taşcı, I. Acute Rhinosinusitis as an Infrequent Cause of Symptomatic Cluster Headache: Report of Seven Cases. J. Oral Facial Pain Headache 2019, 39, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Rose, F.C. Cluster headache and herpes simplex: An association? Br. Med. J. (Clin. Res. Ed.) 1985, 290, 1625–1626. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sacquegna, T.; D’Alessandro, R.; Cortelli, P.; de Carolis, P.; Baldrati, A. Cluster Headache After Herpes Zoster Ophthalmicus. Arch. Neurol. 1982, 39, 384. [Google Scholar] [CrossRef] [PubMed]
- Hakim, S.M. Warfarin for Refractory Chronic Cluster Headache: A Randomized Pilot Study. Headache J. Head Face Pain 2011, 51, 713–725. [Google Scholar] [CrossRef]
- Linehan, J.D.; Kolios, G.; Valatas, V.; Robertson, D.A.F.; Westwick, J. Effect of corticosteroids on nitric oxide production in inflammatory bowel disease: Are leukocytes the site of action? Am. J. Physiol. Liver Physiol. 2005, 288, G261–G267. [Google Scholar] [CrossRef][Green Version]
- Obermann, M.; Nägel, S.; Ose, C.; Sonuc, N.; Scherag, A.; Storch, P.; Gaul, C.; Böger, A.; Kraya, T.; Jansen, J.-P.; et al. Safety and efficacy of prednisone versus placebo in short-term prevention of episodic cluster headache: A multicentre, double-blind, randomised controlled trial. Lancet Neurol. 2021, 20, 29–37. [Google Scholar] [CrossRef]
- Antonaci, F.; Costa, A.; Candeloro, E.; Sjaastad, O.; Nappi, G. Single High-Dose Steroid Treatment in Episodic Cluster Headache. Cephalalgia 2005, 25, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, A.; Wiklund, N.P.; Brundin, L.; Remahl, A.I.M.N. Levels of nitric oxide metabolites in cerebrospinal fluid in cluster headache. Cephalalgia 2010, 30, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Snoer, A.; Lund, N.; Beske, R.; Jensen, R.; Barloese, M. Pre-attack signs and symptoms in cluster headache: Characteristics and time profile. Cephalalgia 2018, 38, 1128–1137. [Google Scholar] [CrossRef]
- Andrée, C.; Barone-Kaganas, I.; Biethahn, S.; Böttger, K.; Dozier, C.; Emmenegger, M.J.; Flügel, D.; Galli, U.; Gantenbein, A.R.; Gobbi, C.; et al. Therapieempfehlungen für Primäre Kopfschmerzen; Schweizerische Kopfwehgesellschaft SKG: Basel, Switzerland, 2019; Available online: https://headache.ch/download/Content_attachments/FileBaseDoc/SKG_Therapieempfehlungen_DE_19_WEB.pdf (accessed on 4 December 2020).
- Pearson, S.M.; Burish, M.J.; Shapiro, R.E.; Yan, Y.; Schor, L.I. Effectiveness of Oxygen and Other Acute Treatments for Cluster Headache: Results From the Cluster Headache Questionnaire, an International Survey. Headache J. Head Face Pain 2019, 59, 235–249. [Google Scholar] [CrossRef]
- Akerman, S.; Holland, P.; Summ, O.; Lasalandra, M.P.; Goadsby, P. A translational in vivo model of trigeminal autonomic cephalalgias: Therapeutic characterization. Brain 2012, 135, 3664–3675. [Google Scholar] [CrossRef]
- Hamel, E. The Biology of Serotonin Receptors: Focus on Migraine Pathophysiology and Treatment. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1999, 26, 2–6. [Google Scholar] [CrossRef]
- Strempel, H. Orcadian cycles of epicritic and protopathic pain threshold. J. Interdiscip. Cycle Res. 1977, 8, 276–280. [Google Scholar] [CrossRef]
- Lee, M.J.; Cho, S.-J.; Park, J.W.; Chu, M.K.; Moon, H.-S.; Chung, P.-W.; Chung, J.-M.; Sohn, J.-H.; Kim, B.-K.; Kim, S.-K.; et al. Temporal changes of circadian rhythmicity in cluster headache. Cephalalgia 2019, 40, 278–287. [Google Scholar] [CrossRef]
- Drummond, P.D.; Lance, J.W. Pathological sweating and flushing accompanying the trigeminal lacrimal reflex in patients with cluster headache and in patients with a confirmed site of cervical sympathetic deficit. Evidence for parasympathetic cross-innervation. Brain 1992, 115, 115. [Google Scholar] [CrossRef]
- Havelius, U. A Horner-like syndrome and cluster headache. What comes first? Acta Ophthalmol. Scand. 2001, 79, 374–375. [Google Scholar] [CrossRef]
- Van Oosterhout, W. Patient with Cluster Headache and Harlequin Sign—Related or Not? Headache J. Head Face Pain 2020, 60, 1761–1766. [Google Scholar] [CrossRef]
- Goadsby, P.; Lambert, G.; Lance, J. Effects of locus coeruleus stimulation on carotid vascular resistance in the cat. Brain Res. 1983, 278, 175–183. [Google Scholar] [CrossRef]
- Ekbom, K.; Greitz, T. Carotid Angiography in Cluster Headache. Acta Radiol. Diagn. 1970, 10, 177–186. [Google Scholar] [CrossRef]
- Drummond, P. Mechanisms of Autonomic Disturbance in the Face During and Between Attacks of Cluster Headache. Cephalalgia 2006, 26, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Straube, A.; Freilinger, T.; Rüther, T.; Padovan, C. Two Cases of Symptomatic Cluster-Like Headache Suggest the Importance of Sympathetic/Parasympathetic Balance. Cephalalgia 2007, 27, 1069–1073. [Google Scholar] [CrossRef]
- Bülbring, E.; Whitteridge, D. The effect of adrenaline on nerve action potentials. J. Physiol. 1941, 99, 201–207. [Google Scholar] [CrossRef]
- Shyu, B.C.; Olausson, B.; Huang, K.H.; Widerström, E.; Andersson, S.A. Effects of sympathetic stimulation on C-fibre responses in rabbit. Acta Physiol. Scand. 1989, 137, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.-C.; Dave, A.S.; Tan, A.Y.; Gholmieh, G.; Zhou, S.; Wang, D.C.; Akingba, A.G.; Fishbein, G.A.; Montemagno, C.; Lin, S.-F.; et al. Circadian variations of stellate ganglion nerve activity in ambulatory dogs. Hear. Rhythm. 2006, 3, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Albertyn, J.; Barry, R.; Odendaal, C.L. Cluster headache and the sympathetic nerve. Headache J. Head Face Pain 2004, 44, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Massin, M.M.; Maeyns, K.; Withofs, N.; Ravet, F.; Gérard, P.; Healy, M.J.R. Circadian rhythm of heart rate and heart rate variability. Arch. Dis. Child. 2000, 83, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, G.; Middleton, B.; Rajaratnam, S.; Stone, B.M.; Thorleifsdottir, B.; Arendt, J.; Dijk, D.-J. Robust circadian rhythm in heart rate and its variability: Influence of exogenous melatonin and photoperiod. J. Sleep Res. 2007, 16, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Nahman-Averbuch, H.; Dayan, L.; Sprecher, E.; Hochberg, U.; Brill, S.; Yarnitsky, D.; Jacob, G. Sex differences in the relationships between parasympathetic activity and pain modulation. Physiol. Behav. 2016, 154, 40–48. [Google Scholar] [CrossRef]
- Meineri, P.; Pellegrino, G.; Rosso, M.G.; Grasso, E. Systemic autonomic involvement in episodic cluster headache: A comparison between active and remission periods. J. Headache Pain 2005, 6, 240–243. [Google Scholar] [CrossRef]
- Waldenlind, E.; Gustafsson, S.A.; Ekbom, K.; Wetterberg, L. Circadian secretion of cortisol and melatonin in cluster headache during active cluster periods and remission. J. Neurol. Neurosurg. Psychiatry 1987, 50, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Chazot, G.; Claustrat, B.; Brun, J.; Jordan, D.; Sassolas, G.; Schott, B. A Chronobiological Study of Melatonin, Cortisol Growth Hormone and Prolactin Secretion in Cluster Headache. Cephalalgia 1984, 4, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gharaghozlu, N.; Ahmadi, R.; Khakpour, B.; Alvany, A.; Keshavarz, X. Effects of Testosterone on Pain Threshold. In Proceedings of the International Conference on Chemical, Agricultural and Medical Sciences, Antalaya, Turkey, 2–3 May 2014. [Google Scholar]
- Gkika, D.; Lolignier, S.; Grolez, G.P.; Bavencoffe, A.; Shapovalov, G.; Gordienko, D.; Kondratskyi, A.; Meleine, M.; Prival, L.; Chapuy, E.; et al. Testosterone-androgen receptor: The steroid link inhibiting TRPM8-mediated cold sensitivity. FASEB J. 2020, 34, 7483–7499. [Google Scholar] [CrossRef]
- Kobayashi, K.; Fukuoka, T.; Obata, K.; Yamanaka, H.; Dai, Y.; Tokunaga, A.; Noguchi, K. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J. Comp. Neurol. 2005, 493, 596–606. [Google Scholar] [CrossRef]
- Waldenlind, E.; Gustafsson, S.A. Prolactin in Cluster Headache: Diurnal Secretion, Response to Thyrotropin-Releasing Hormone, and Relation to Sex Steroids and Gonadotropins. Cephalalgia 1987, 7, 43–54. [Google Scholar] [CrossRef]
- Léoné, M.; Patruno, G.; Vescovi, A.; Bussone, G. Neuroendocrine Dysfunction in Cluster Headache. Cephalalgia 1990, 10, 235–239. [Google Scholar] [CrossRef]
- Yang, F.-C.; Chou, K.-H.; Fuh, J.-L.; Lee, P.-L.; Lirng, J.-F.; Lin, Y.-Y.; Lin, C.-P.; Wang, S.-J. Altered hypothalamic functional connectivity in cluster headache: A longitudinal resting-state functional MRI study. J. Neurol. Neurosurg. Psychiatry 2014, 86, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Morelli, N.; Pesaresi, I.; Cafforio, G.; Maluccio, M.R.; Gori, S.; Di Salle, F.; Murri, L. Functional magnetic resonance imaging in episodic cluster headache. J. Headache Pain 2008, 10, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Franzini, A.; Broggi, G.; Bussone, G. Hypothalamic stimulation for intractable cluster headache: Long-term experience. Neurology 2006, 67, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Seijo-Fernandez, F.; Saiz, A.; Santamarta, E.; Nader, L.; Alvarez-Vega, M.A.; Lozano, B.; Seijo, E.; Barcia, J.A. Long-Term Results of Deep Brain Stimulation of the Mamillotegmental Fasciculus in Chronic Cluster Headache. Ster. Funct. Neurosurg. 2018, 96, 215–222. [Google Scholar] [CrossRef]
- Nowacki, A.; Schober, M.; Nader, L.; Saryyeva, A.; Nguyen, T.A.K.; Green, A.L.; Pollo, C.; Krauss, J.K.; Fontaine, D.; Aziz, T.Z. DBS for chronic cluster headache: Meta-analysis of individual patient data. Ann. Neurol. 2020, 88, 956–969. [Google Scholar] [CrossRef]
- Ferguson, A.V.; Latchford, K.J.; Samson, W.K. The paraventricular nucleus of the hypothalamus—A potential target for integrative treatment of autonomic dysfunction. Expert Opin. Ther. Targets 2008, 12, 717–727. [Google Scholar] [CrossRef]
- Li, C.; Fitzgerald, M.E.; LeDoux, M.S.; Gong, S.; Ryan, P.; Del Mar, N.; Reiner, A. Projections from the hypothalamic paraventricular nucleus and the nucleus of the solitary tract to prechoroidal neurons in the superior salivatory nucleus: Pathways controlling rodent choroidal blood flow. Brain Res. 2010, 1358, 123–139. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.-M.; Yang, Y.; Liu, W.-Y.; Song, C.-Y.; Lin, B.-C. Investigating the Role of Hypothalamic Paraventricular Nucleus in Nociception of the Rat. Int. J. Neurosci. 2008, 118, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Shen, G. In the rat, endogenous nitric oxide modulates the response of the hypothalamic-pituitary-adrenal axis to interleukin-1 beta, vasopressin, and oxytocin. J. Neurosci. 1994, 14, 1985–1993. [Google Scholar] [CrossRef]
- Patel, K.P. Role of Paraventricular Nucleus in Mediating Sympathetic Outflow in Heart Failure. Hear. Fail. Rev. 2000, 5, 73–86. [Google Scholar] [CrossRef]
- Hofman, M.A.; Swaab, D.F. Seasonal changes in the suprachiasmatic nucleus of man. Neurosci. Lett. 1992, 139, 257–260. [Google Scholar] [CrossRef][Green Version]
- Lee, H.S.; Billings, H.J.; Lehman, M.N. The Suprachiasmatic Nucleus: A Clock of Multiple Components. J. Biol. Rhythm. 2003, 18, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Li, J.; Tang, K. The Paraventricular Nucleus of the Hypothalamus: Development, Function, and Human Diseases. Endocrinology 2018, 159, 3458–3472. [Google Scholar] [CrossRef]
- Lund, N.; Petersen, A.; Snoer, A.; Jensen, R.H.; Barloese, M. Cluster headache is associated with unhealthy lifestyle and lifestyle-related comorbid diseases: Results from the Danish Cluster Headache Survey. Cephalalgia 2019, 39, 254–263. [Google Scholar] [CrossRef]
- Woo, A.K. Depression and Anxiety in Pain. Rev. Pain 2010, 4, 8–12. [Google Scholar] [CrossRef]
- Schuh-Hofer, S.; Wodarski, R.; Pfau, D.B.; Caspani, O.; Magerl, W.; Kennedy, J.D.; Treede, R.-D. One night of total sleep deprivation promotes a state of generalized hyperalgesia: A surrogate pain model to study the relationship of insomnia and pain. Pain 2013, 154, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Sardi, N.F.; Lazzarim, M.K.; Guilhen, V.A.; Marcílio, R.S.; Natume, P.S.; Watanabe, T.C.; Lima, M.M.; Tobaldini, G.; Fischer, L. Chronic sleep restriction increases pain sensitivity over time in a periaqueductal gray and nucleus accumbens dependent manner. Neuropharmacology 2018, 139, 52–60. [Google Scholar] [CrossRef]
- Liang, J.-F.; Chen, Y.-T.; Fuh, J.-L.; Li, S.-Y.; Liu, C.-J.; Chen, T.-J.; Tang, C.-H.; Wang, S.-J. Cluster headache is associated with an increased risk of depression: A nationwide population-based cohort study. Cephalalgia 2013, 33, 182–189. [Google Scholar] [CrossRef]
- Schytz, H.W.; Barløse, M.; Guo, S.; Selb, J.; Caparso, A.; Jensen, R.; Ashina, M. Experimental activation of the sphenopalatine ganglion provokes cluster-like attacks in humans. Cephalalgia 2013, 33, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Lignani, G.; Baldelli, P.; Marra, V. Homeostatic Plasticity in Epilepsy. Front. Cell. Neurosci. 2020, 14, 197. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, K. Nitrolglycerin as a provocative agent in cluster headache. Arch. Neurol. 1968, 19, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, S.; Nigri, A.; Demichelis, G.; Pinardi, C.; Chiapparini, L.; Giani, L.; Cecchini, A.P.; Leone, M. Understanding Cluster Headache Using Magnetic Resonance Imaging. Front. Neurol. 2020, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.Y.; Goadsby, P.J. Cluster headache pathophysiology—Insights from current and emerging treatments. Nat. Rev. Neurol. 2021, 17, 308–324. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pohl, H.; Sandor, P.S.; Michels, L.; Gantenbein, A.R. Cluster Headache Pathophysiology—A Disorder of Network Excitability? Clin. Transl. Neurosci. 2021, 5, 16. https://doi.org/10.3390/ctn5020016
Pohl H, Sandor PS, Michels L, Gantenbein AR. Cluster Headache Pathophysiology—A Disorder of Network Excitability? Clinical and Translational Neuroscience. 2021; 5(2):16. https://doi.org/10.3390/ctn5020016
Chicago/Turabian StylePohl, Heiko, Peter S. Sandor, Lars Michels, and Andreas R. Gantenbein. 2021. "Cluster Headache Pathophysiology—A Disorder of Network Excitability?" Clinical and Translational Neuroscience 5, no. 2: 16. https://doi.org/10.3390/ctn5020016
APA StylePohl, H., Sandor, P. S., Michels, L., & Gantenbein, A. R. (2021). Cluster Headache Pathophysiology—A Disorder of Network Excitability? Clinical and Translational Neuroscience, 5(2), 16. https://doi.org/10.3390/ctn5020016