
Driven Transport of Dilute Polymer Solutions through Porous Media Comprising Interconnected Cavities


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
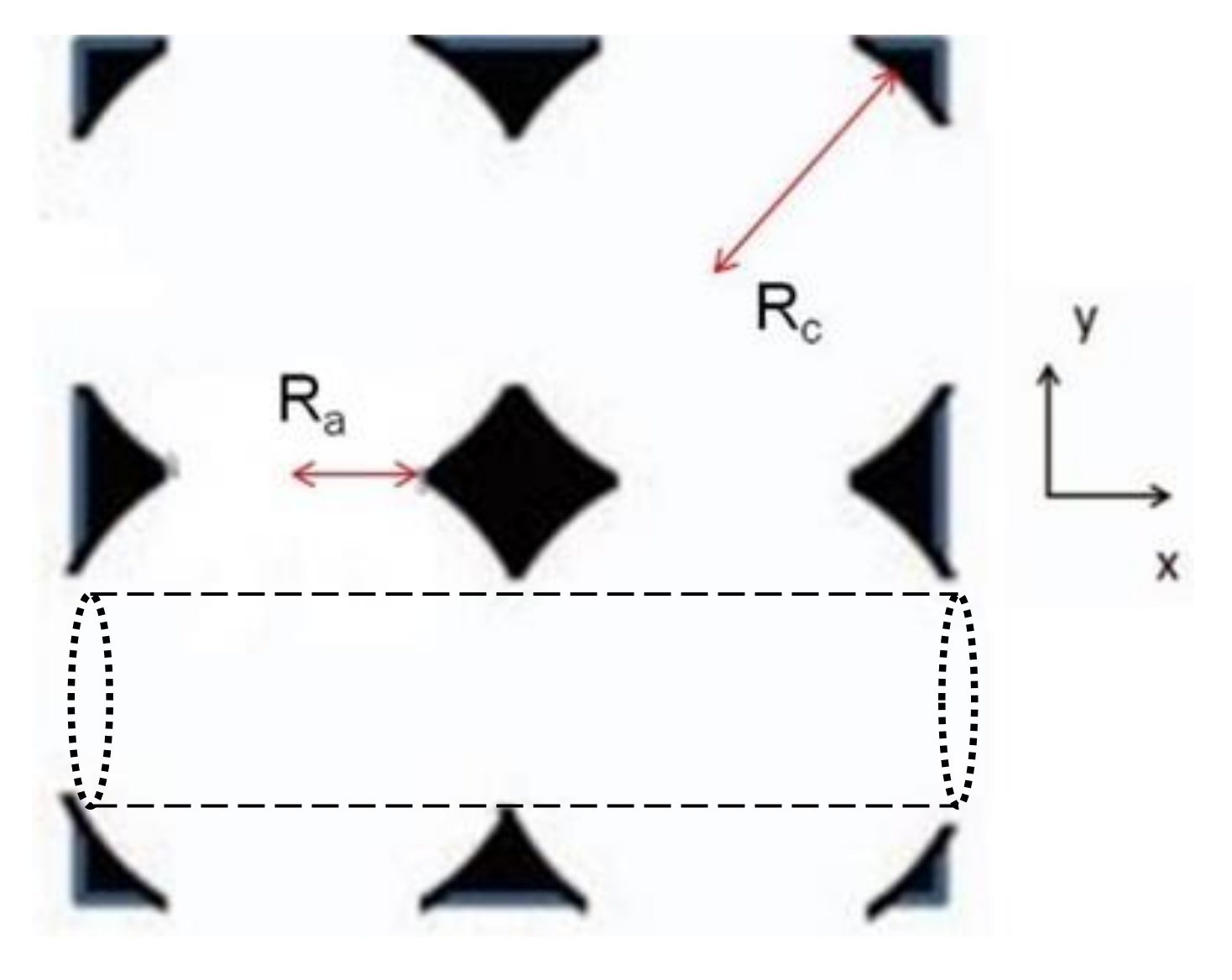
2. Methods
3. Results and Discussion
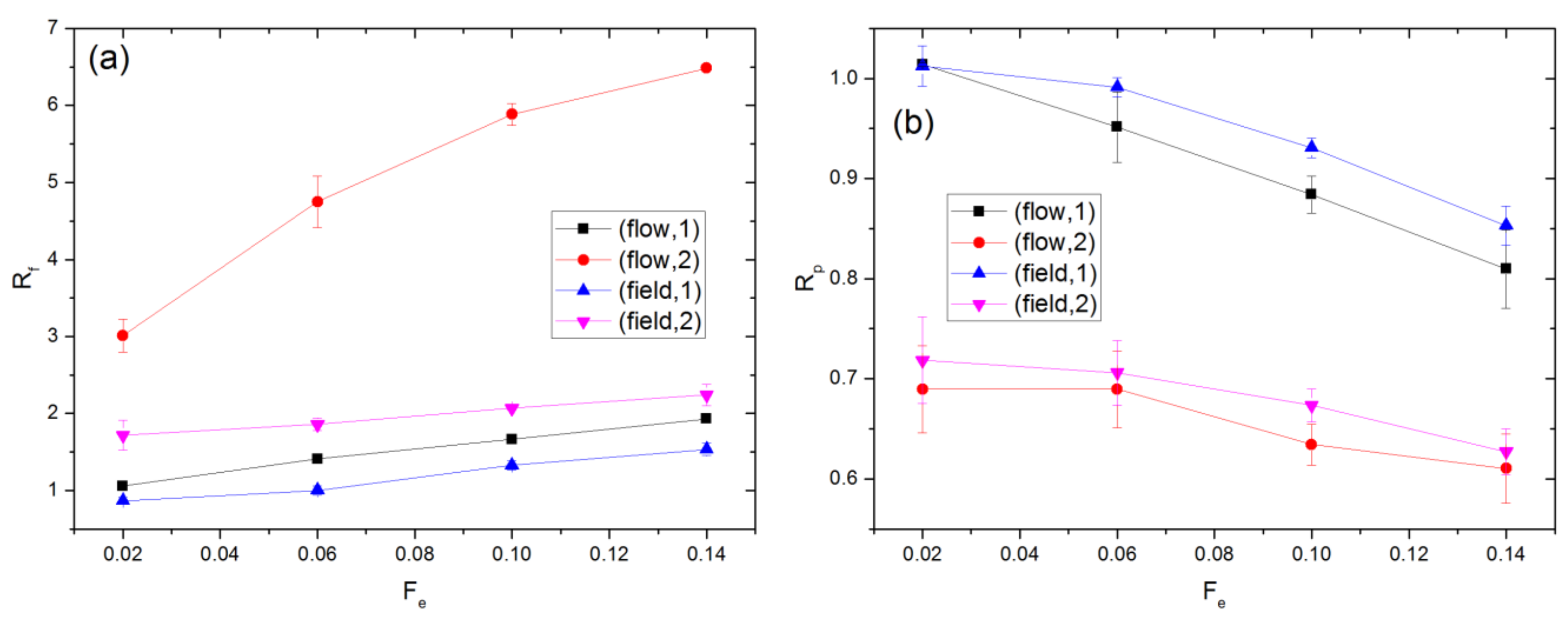
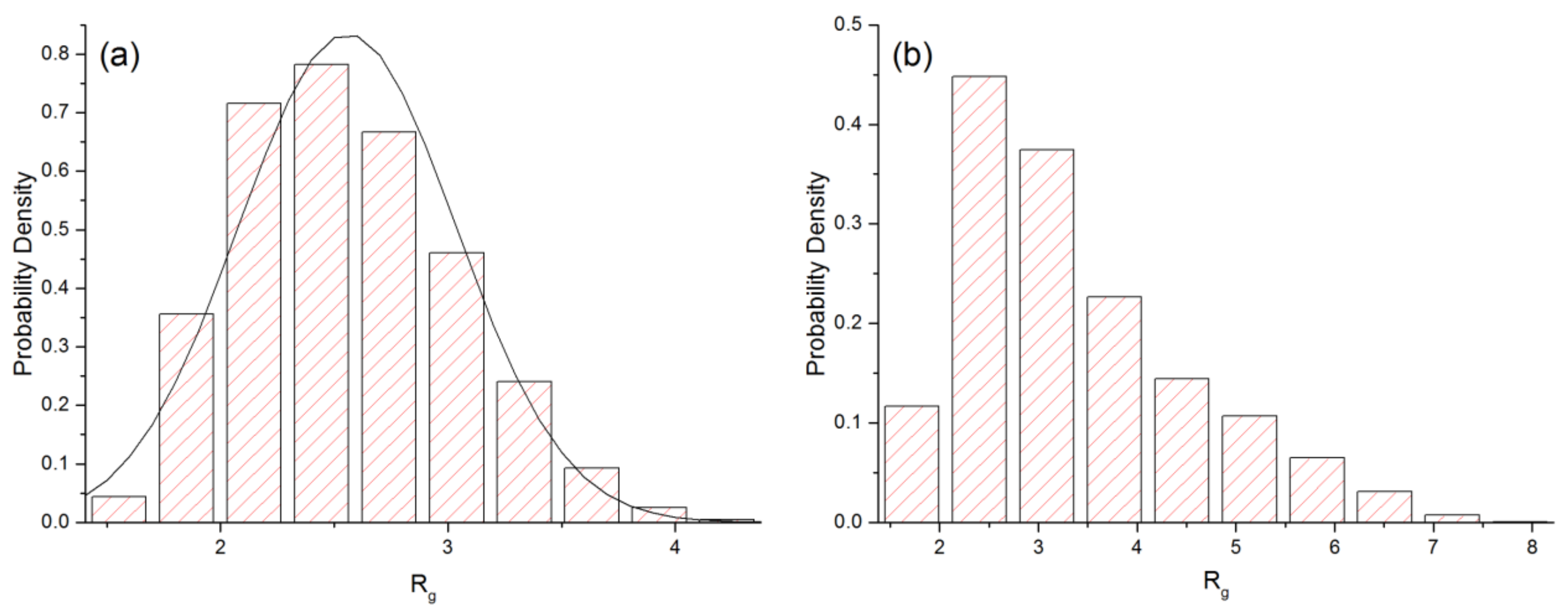
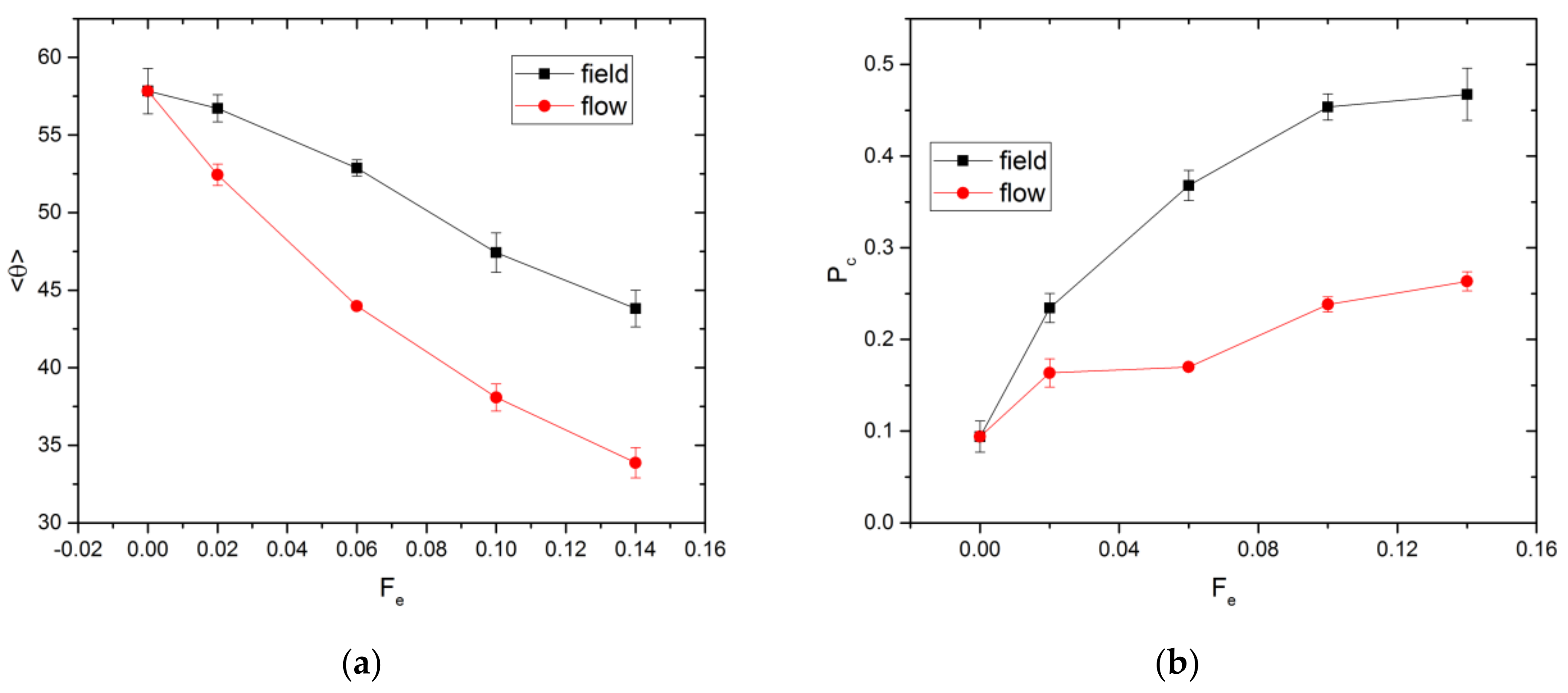
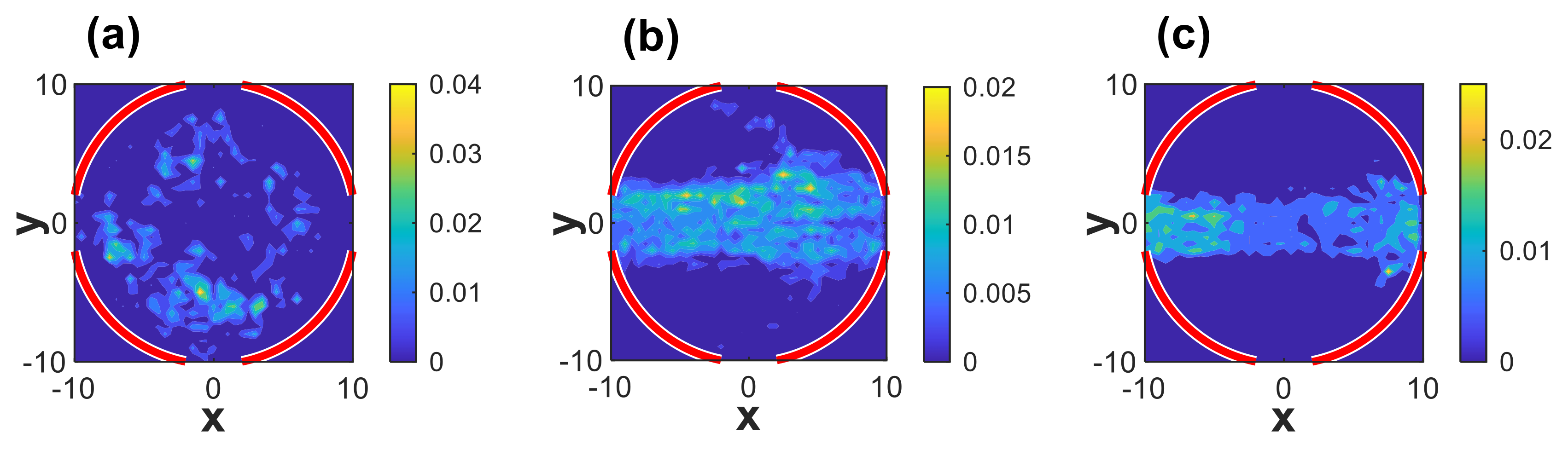
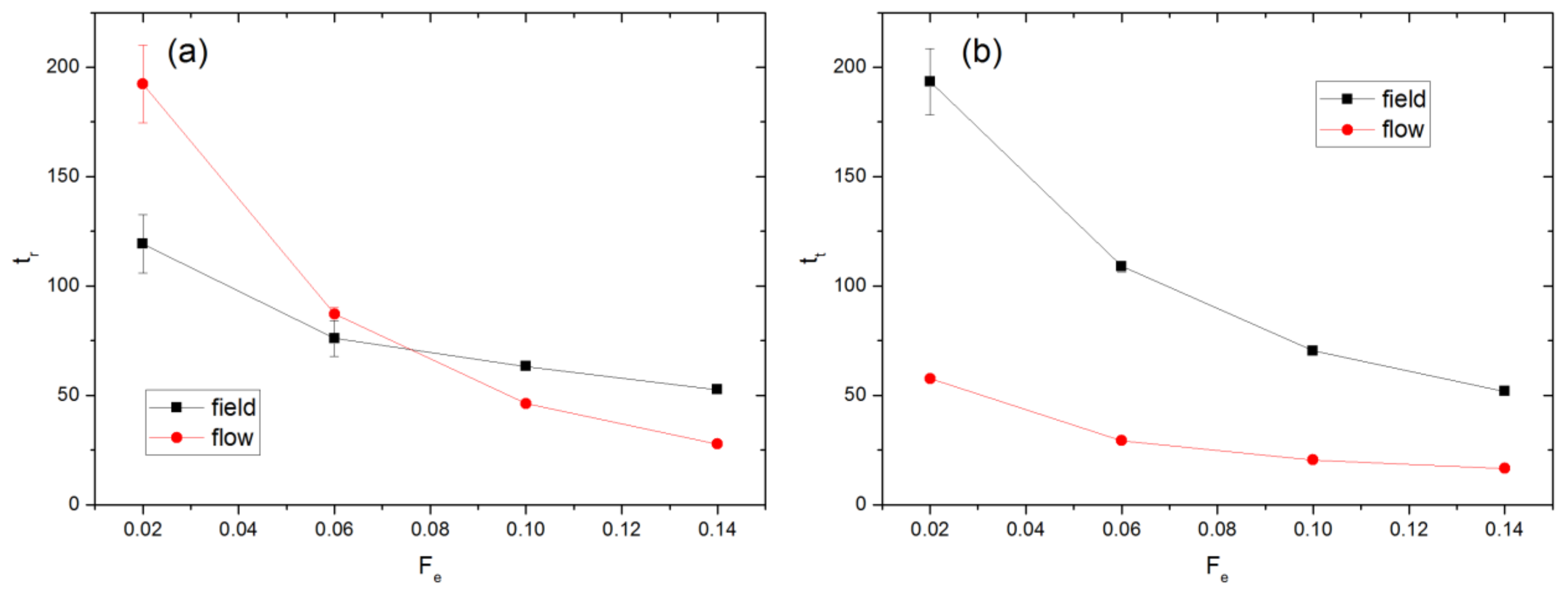
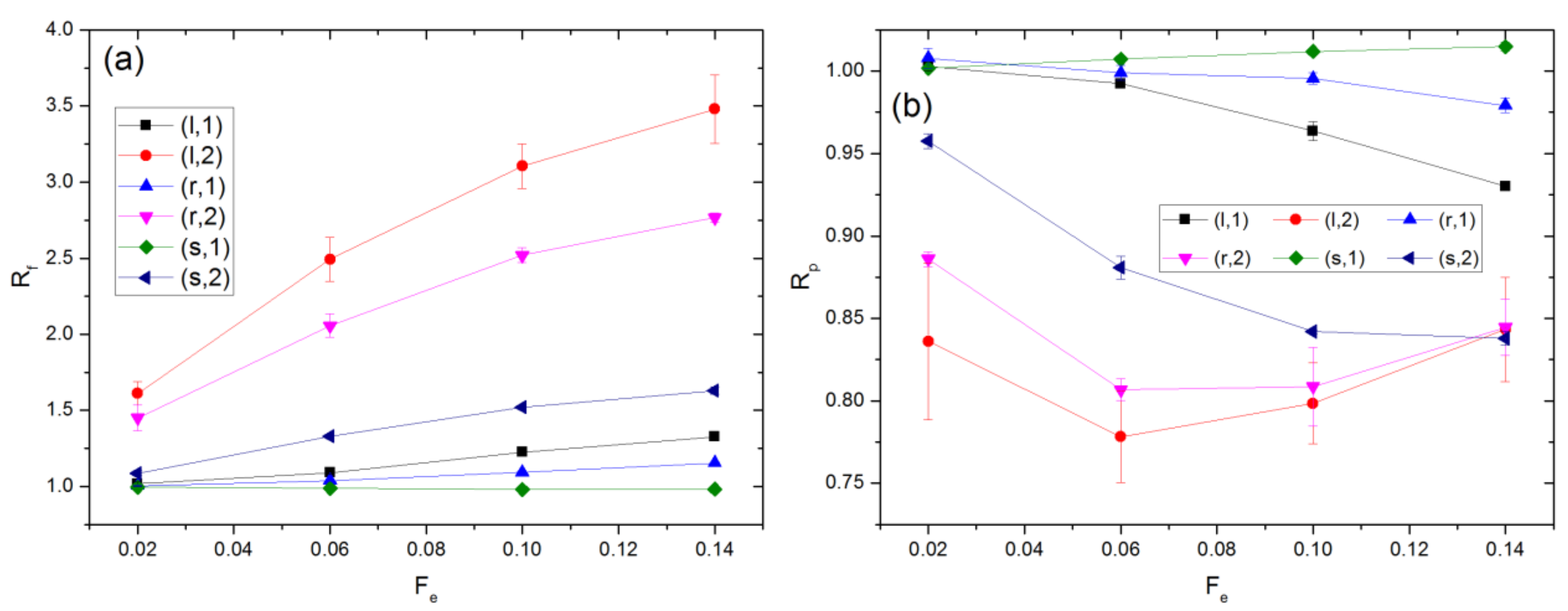
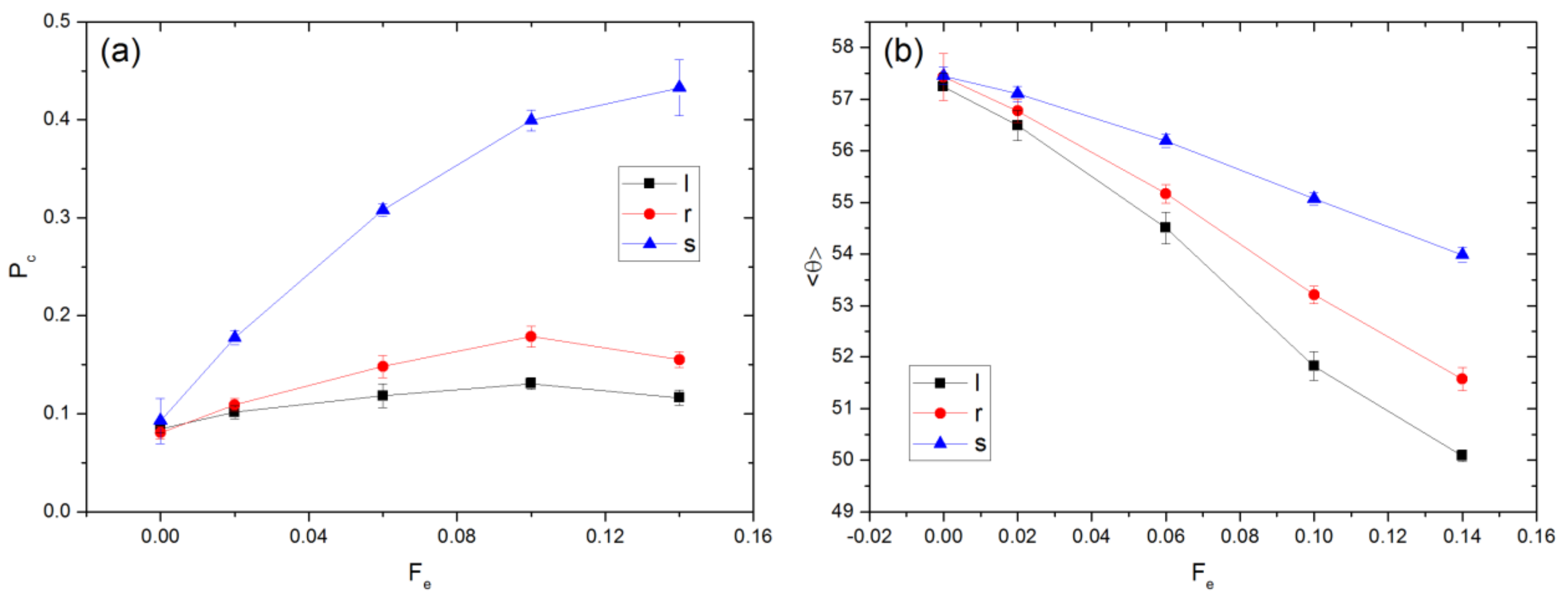
3.1. Linear Polymers
3.2. Effect of Polymer Architecture
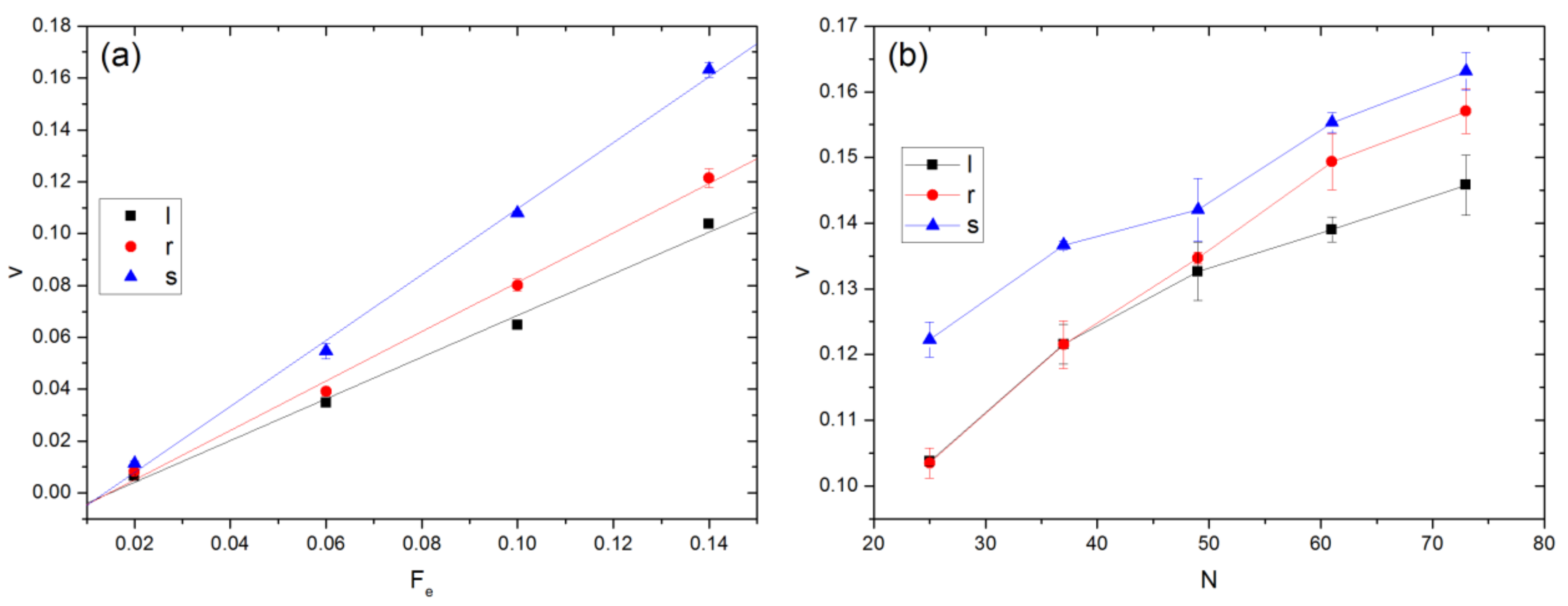
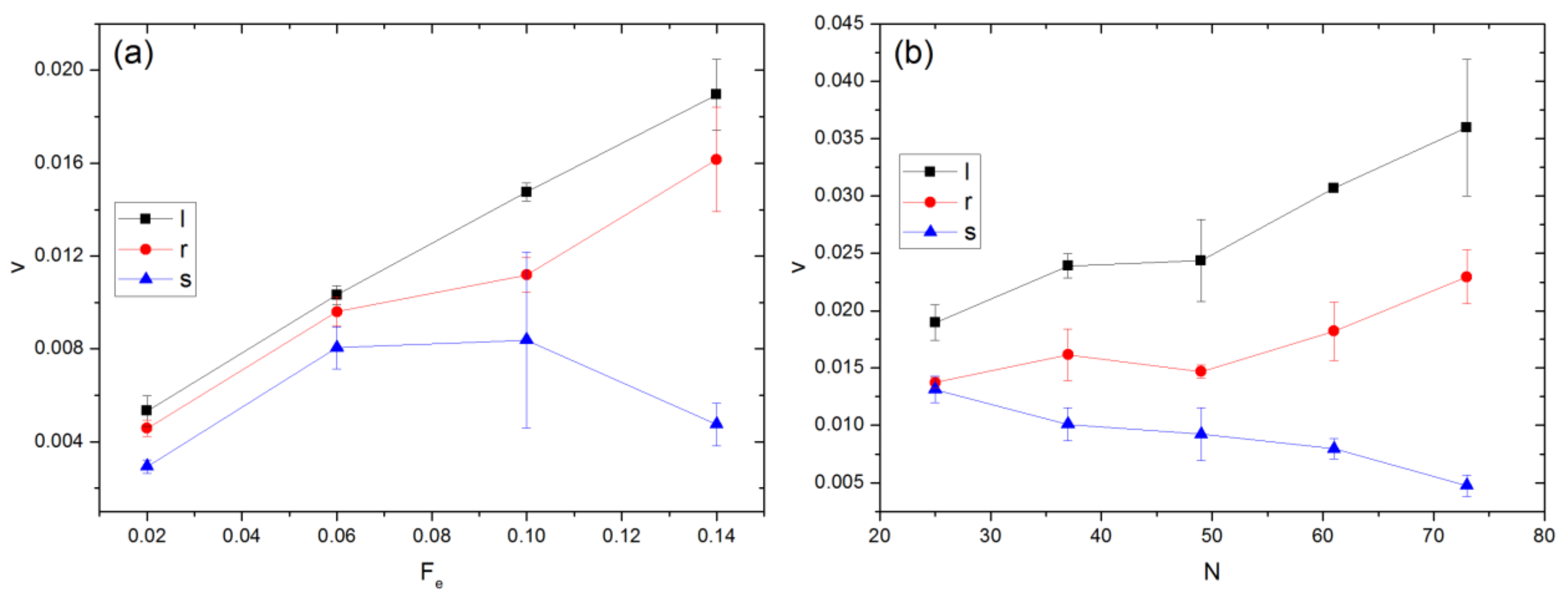
3.3. Velocity of COM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abidin, A.Z.; Puspasari, T.; Nugroho, W.A. Polymers for Enhanced Oil Recovery Technology. Procedia Chem. 2012, 4, 11–16. [Google Scholar] [CrossRef]
- Viovy, J.-L. Electrophoresis of DNA and other polyelectrolytes: Physical mechanisms. Rev. Mod. Phys. 2000, 72, 813–872. [Google Scholar] [CrossRef]
- Dorfman, K.D. DNA electrophoresis in microfabricated devices. Rev. Mod. Phys. 2010, 82, 2903–2947. [Google Scholar] [CrossRef]
- Dorfman, K.D.; King, S.B.; Olson, D.W.; Thomas, J.D.P.; Tree, D.R. Beyond gel electrophoresis: Microfluidic separations, fluorescence burst analysis, and DNA stretching. Chem. Rev. 2013, 113, 2584–2667. [Google Scholar] [CrossRef]
- Graham, M.D. Fluid Dynamics of Dissolved Polymer Molecules in Confined Geometries. Annu. Rev. Fluid Mech. 2011, 43, 273–298. [Google Scholar] [CrossRef]
- DeLong, C.D.; Hoagland, D.A. Imaging of Individual Polymers Undergoing Flow in a Bed of Small Spheres. Macromolecules 2008, 41, 4887–4894. [Google Scholar] [CrossRef]
- Schiller, U.D.; Krüger, T.; Henrich, O. Mesoscopic modelling and simulation of soft matter. Soft Matter 2017, 14, 9–26. [Google Scholar] [CrossRef]
- Slater, G.W.; Holm, C.; Chubynsky, M.V.; de Haan, H.W.; Dubé, A.; Grass, K.; Hickey, O.A.; Kingsburry, C.; Sean, D.; Shendruk, T.N.; et al. Modeling the separation of macromolecules: A review of current computer simulation methods. Electrophoresis 2009, 30, 792–818. [Google Scholar] [CrossRef]
- Ding, M.; Duan, X.; Shi, T. Flow-induced translocation of star polymers through a nanopore. Soft Matter 2016, 12, 2851–2857. [Google Scholar] [CrossRef]
- Ding, M.; Chen, Q.; Duan, X.; Shi, T. Flow-Driven Translocation of a Diblock Copolymer through a Nanopore. J. Phys. Chem. B 2019, 123, 8848–8852. [Google Scholar] [CrossRef]
- Fan, X.; Phan-Thien, N.N.; Yong, N.T.; Wu, X.; Xu, D. Microchannel flow of a macromolecular suspension. Phys. Fluids 2003, 15, 11–21. [Google Scholar] [CrossRef]
- Fan, X.; Phan-Thien, N.; Chen, S.; Wu, X.; Ng, T.Y. Simulating flow of DNA suspension using dissipative particle dynamics. Phys. Fluids 2006, 18, 063102. [Google Scholar] [CrossRef]
- Millan, J.A.; Jiang, W.; Laradji, M.; Wang, Y. Pressure Driven Flow of Polymer Solutions in Nanoscale Slit Pores. J. Chem. Phys. 2006, 126, 1–11. [Google Scholar] [CrossRef]
- Millan, J.A.; Laradji, M. Cross-Stream Migration of Driven Polymer Solutions in Nanoscale Channels: A Numerical Study with Generalized Dissipative Particle Dynamics. Macromolecules 2009, 42, 803–810. [Google Scholar] [CrossRef]
- Danioko, S.; Laradji, M. Tumbling, stretching and cross-stream migration of polymers in rectilinear shear flow from dissipative particle dynamics simulations. Phys. A Stat. Mech. Appl. 2012, 391, 3379–3391. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Wang, Y.; Sun, Z.; An, L. Transport of star-branched polymers in nanoscale pipe channels simulated with dissipative particle dynamics simulation. Macromolecules 2010, 43, 5896–5903. [Google Scholar] [CrossRef]
- Posel, Z.; Svoboda, M.; Colina, C.M.; Lísal, M. Flow and aggregation of rod-like proteins in slit and cylindrical pores coated with polymer brushes: An insight from dissipative particle dynamics. Soft Matter 2017, 13, 1634–1645. [Google Scholar] [CrossRef]
- Hernandez-Ortiz, J.P.; Ma, H.; de Pablo, J.J.; Graham, M.D. Concentration distributions during flow of confined flowing polymer solutions at finite concentration: Slit and grooved channel. Korea Aust. Rheol. J. 2008, 20, 143–152. [Google Scholar]
- Jiang, L.; Larson, R.G. Multiscale modeling of polymer flow-induced migration and size separation in a microfluidic contraction flow. J. Nonnewton. Fluid Mech. 2014, 211, 84–98. [Google Scholar] [CrossRef]
- Muthukumar, M.; Baumgartner, A. Effects of entropic barriers on polymer dynamics. Macromolecules 1989, 22, 1937–1941. [Google Scholar] [CrossRef]
- Muthukumar, M.; Baumgärtner, A. Diffusion of a Polymer Chain in Random Media. Macromolecules 1989, 22, 1941–1946. [Google Scholar] [CrossRef]
- Nykypanchuk, D.; Strey, H.H.; Hoagland, D. Brownian Motion of DNA Confined Within a Two-Dimensional Array. Science 2002, 297, 987–990. [Google Scholar] [CrossRef]
- Nykypanchuk, D.; Strey, H.H.; Hoagland, D.A. Single molecule visualizations of polymer partitioning within model pore geometries. Macromolecules 2005, 38, 145–150. [Google Scholar] [CrossRef]
- Nykypanchuk, D.; Hoagland, D.A.; Strey, H.H. Diffusion of circular DNA in two-dimensional cavity arrays. ChemPhysChem 2009, 10, 2847–2851. [Google Scholar] [CrossRef]
- Zeng, Y.; Harrison, D.J. Confinement effects on electromigration of long DNA molecules in an ordered cavity array. Electrophoresis 2006, 27, 3747–3752. [Google Scholar] [CrossRef]
- Chen, S.B. Driven transport of particles in 3D ordered porous media. J. Chem. Phys. 2013, 139, 074904. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative Particle Dynamics: Bridging the Gap between Atomistic and Mesoscopic Simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Jiang, W.; Huang, J.; Wang, Y.; Laradji, M. Hydrodynamic interaction in polymer solutions simulated with dissipative particle dynamics. J. Chem. Phys. 2007, 126, 044901. [Google Scholar] [CrossRef]
- Nardai, M.M.; Zifferer, G. Simulation of dilute solutions of linear and star-branched polymers by dissipative particle dynamics. J. Chem. Phys. 2009, 131, 124903. [Google Scholar] [CrossRef]
- Liu, M.B.; Liu, G.R.; Zhou, L.W.; Chang, J.Z. Dissipative Particle Dynamics (DPD): An Overview and Recent Developments. Arch. Comput. Methods Eng. 2014, 22, 529–556. [Google Scholar] [CrossRef]
- Xu, Z.; Meakin, P. A phase-field approach to no-slip boundary conditions in dissipative particle dynamics and other particle models for fluid flow in geometrically complex confined systems. J. Chem. Phys. 2009, 130, 234103. [Google Scholar] [CrossRef] [PubMed]
- Duong-Hong, D.; Phan-Thien, N.; Fan, X. An implementation of no-slip boundary conditions in DPD. Comput. Mech. 2004, 35, 24–29. [Google Scholar] [CrossRef]
- Li, Z.; Bian, X.; Tang, Y.H.; Karniadakis, G.E. A dissipative particle dynamics method for arbitrarily complex geometries. J. Comput. Phys. 2018, 355, 534–547. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Xiao, M.; Wang, R.; Chen, Y.L. Conformation-dependent translocation of a star polymer through a nanochannel. Biomicrofluidics 2014, 8, 054107. [Google Scholar] [CrossRef]
- Kawale, D.; Bouwman, G.; Sachdev, S.; Zitha, P.L.J.; Kreutzer, M.T.; Rossen, W.R.; Boukany, P.E. Polymer conformation during flow in porous media. Soft Matter 2017, 13, 8745–8755. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Plimpton, S. LAMMPS Molecular Dynamics Simulator. Available online: http://lammps.sandia.gov/ (accessed on 1 January 2019).
- Topgaard, D. Director orientations in lyotropic liquid crystals: Diffusion MRI mapping of the Saupe order tensor. Phys. Chem. Chem. Phys. 2016, 18, 8545–8553. [Google Scholar] [CrossRef]
- Srivastva, D.; Nikoubashman, A. Flow behavior of chain and star polymers and their mixtures. Polymers 2018, 10, 599. [Google Scholar] [CrossRef]
- Han, J.; Turner, S.; Craighead, H. Entropic Trapping and Escape of Long DNA Molecules at Submicron Size Constriction. Phys. Rev. Lett. 1999, 83, 1688–1691. [Google Scholar] [CrossRef]
- Moeendarbary, E.; Ng, T.Y.; Pan, H.; Lam, K.Y. Migration of DNA molecules through entropic trap arrays: A dissipative particle dynamics study. Microfluid. Nanofluidics 2010, 8, 243–254. [Google Scholar] [CrossRef]
- Han, A.J.; Craighead, H.G. Separation of Long DNA Molecules in a Microfabricated Entropic Trap Array. Science 2000, 288, 1026–1029. [Google Scholar] [CrossRef]
- Han, J.; Craighead, H.G. Characterization and optimization of an entropic trap for DNA separation. Anal. Chem. 2002, 74, 394–401. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagarajan, K.; Chen, S.B. Driven Transport of Dilute Polymer Solutions through Porous Media Comprising Interconnected Cavities. Colloids Interfaces 2021, 5, 22. https://doi.org/10.3390/colloids5020022
Nagarajan K, Chen SB. Driven Transport of Dilute Polymer Solutions through Porous Media Comprising Interconnected Cavities. Colloids and Interfaces. 2021; 5(2):22. https://doi.org/10.3390/colloids5020022
Chicago/Turabian StyleNagarajan, Karthik, and Shing Bor Chen. 2021. "Driven Transport of Dilute Polymer Solutions through Porous Media Comprising Interconnected Cavities" Colloids and Interfaces 5, no. 2: 22. https://doi.org/10.3390/colloids5020022
APA StyleNagarajan, K., & Chen, S. B. (2021). Driven Transport of Dilute Polymer Solutions through Porous Media Comprising Interconnected Cavities. Colloids and Interfaces, 5(2), 22. https://doi.org/10.3390/colloids5020022