
Carbonated 3D-Printable Polymer Composite for Thermo-Mechanically Stable Applications


Abstract
:
1. Introduction
2. Material and Methods
2.1. Sample Preparation
2.2. Sample Analysis
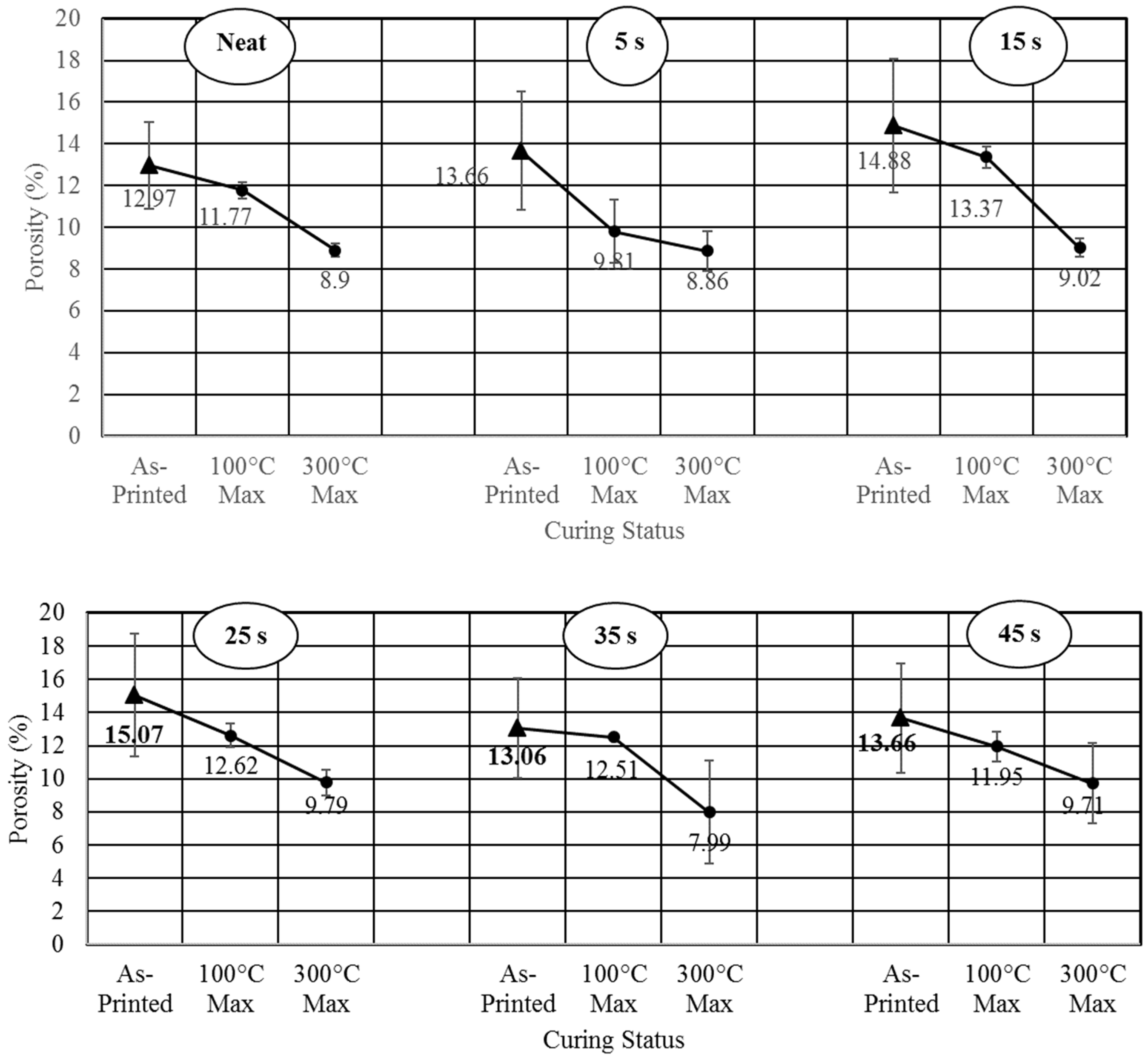
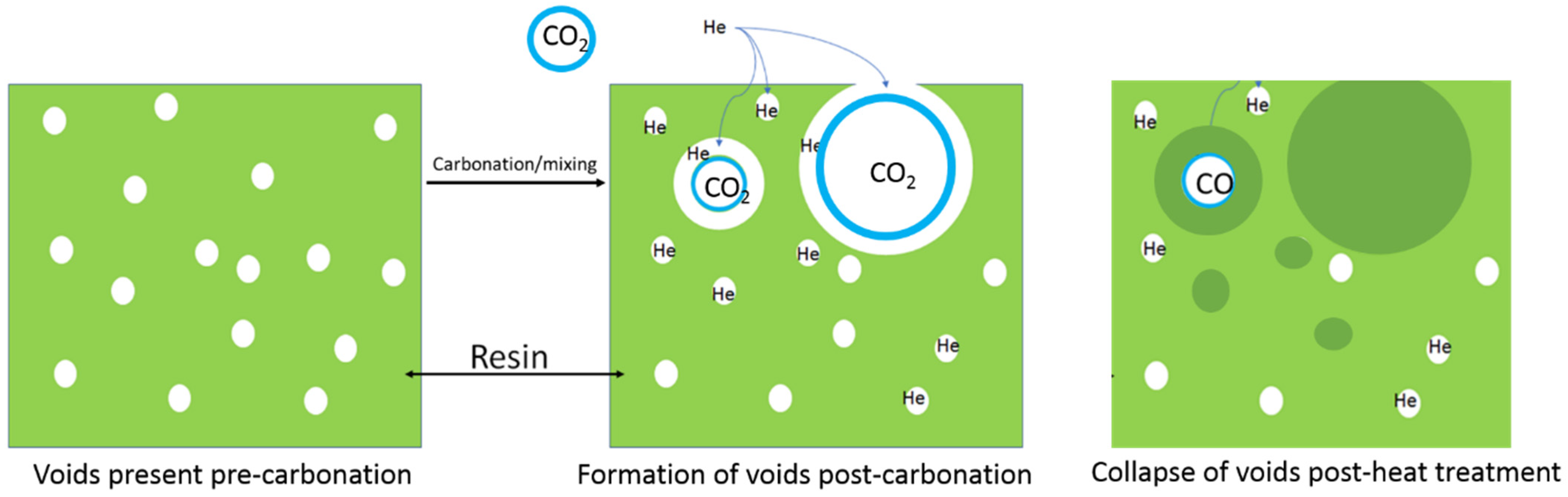
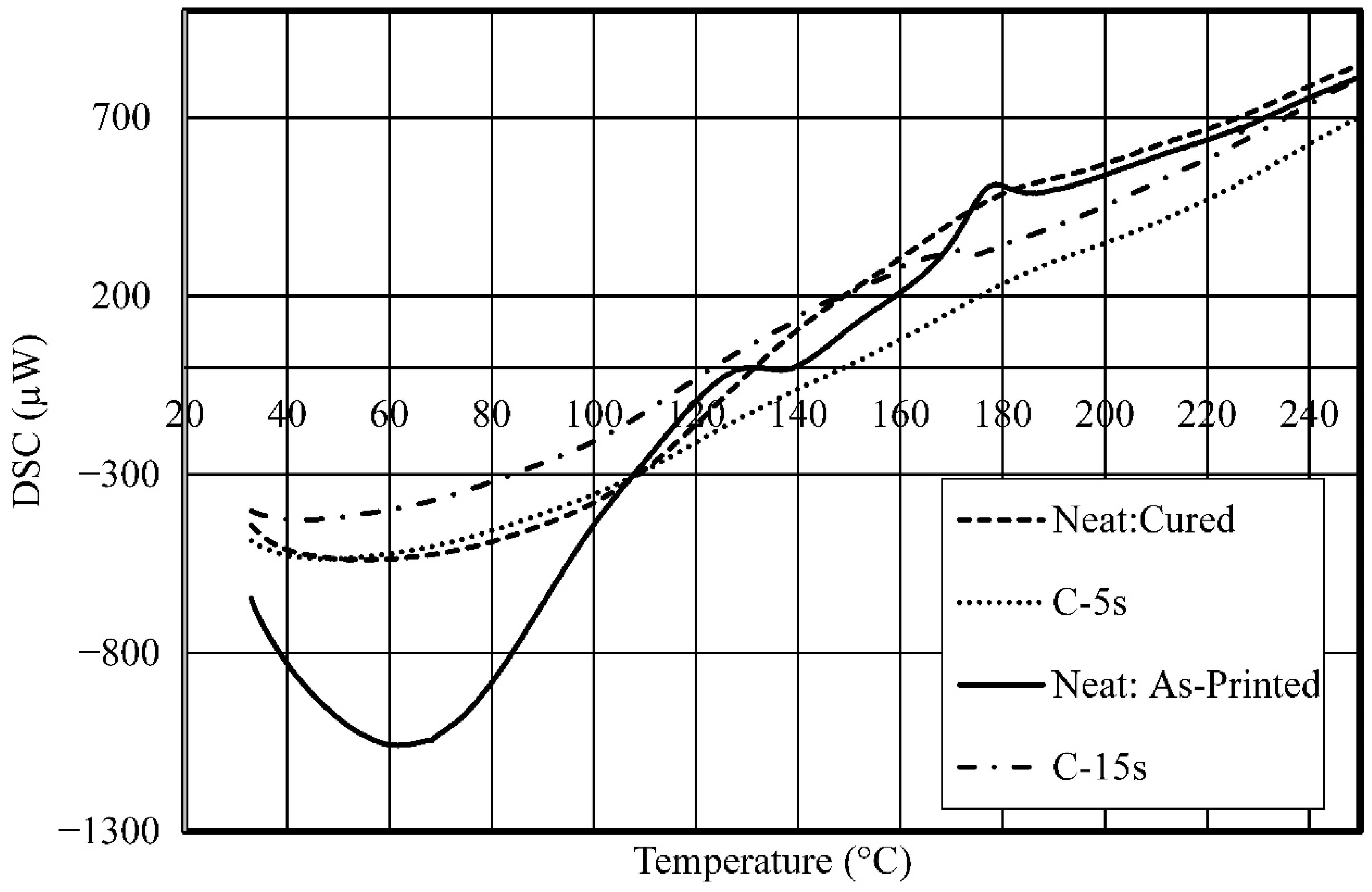
3. Results and Discussion
4. Conclusions
- Synthesizing a carbonated polymer composite material using a method akin to liquid carbonation
- Additive manufacturing of the carbonated polymer composite proved possible using the digital light projection (DLP) 3D-printing technique
- Characterization revealed that the CO2 gas molecule physically behaves as a solid particulate filler. This observation gives rise to the consideration of utilizing an assortment of other gas molecules and mixtures with unique properties as polymer enhancement fillers, thereby expanding the selection of polymer composite materials and manufacturing processes.
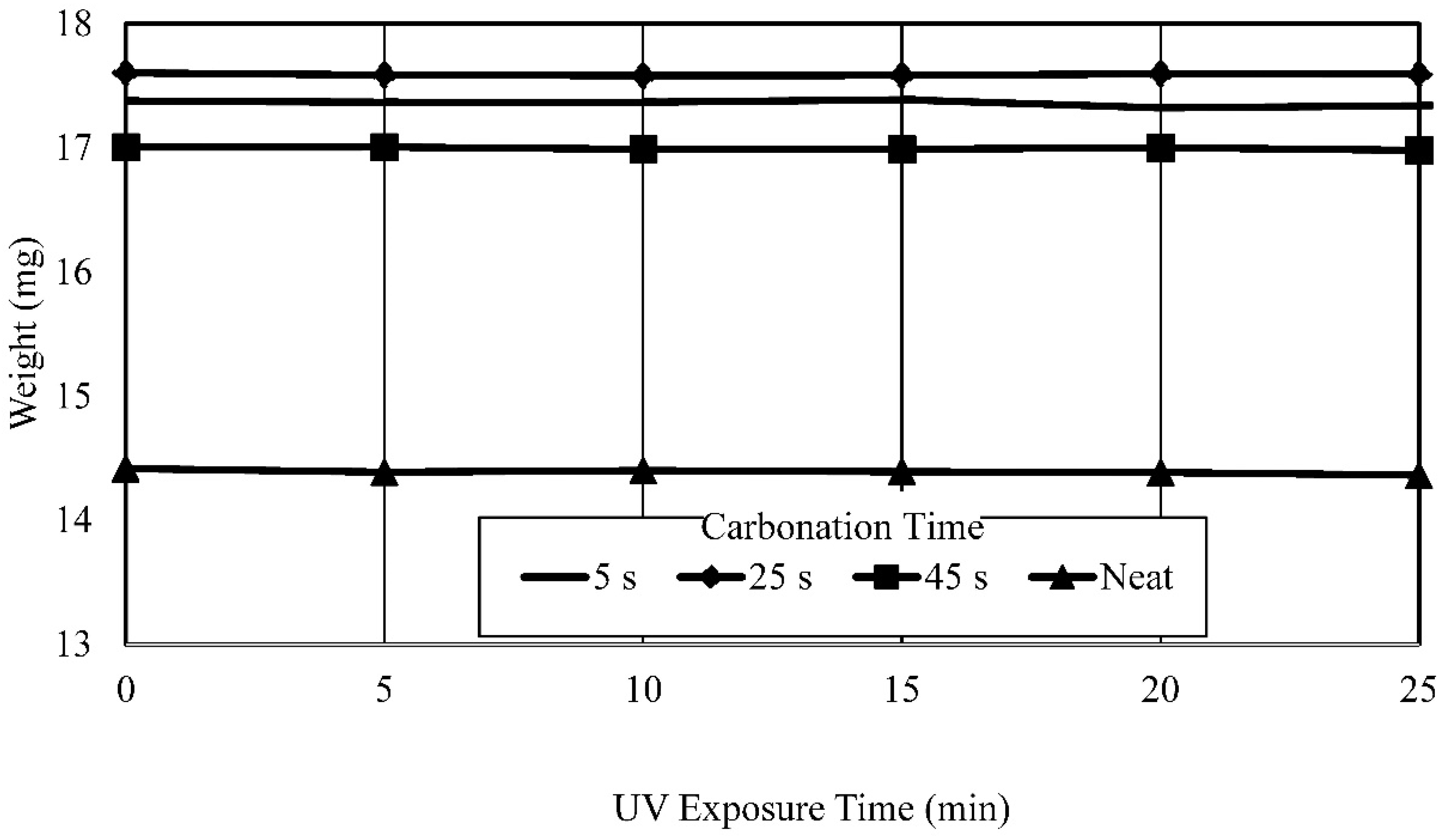
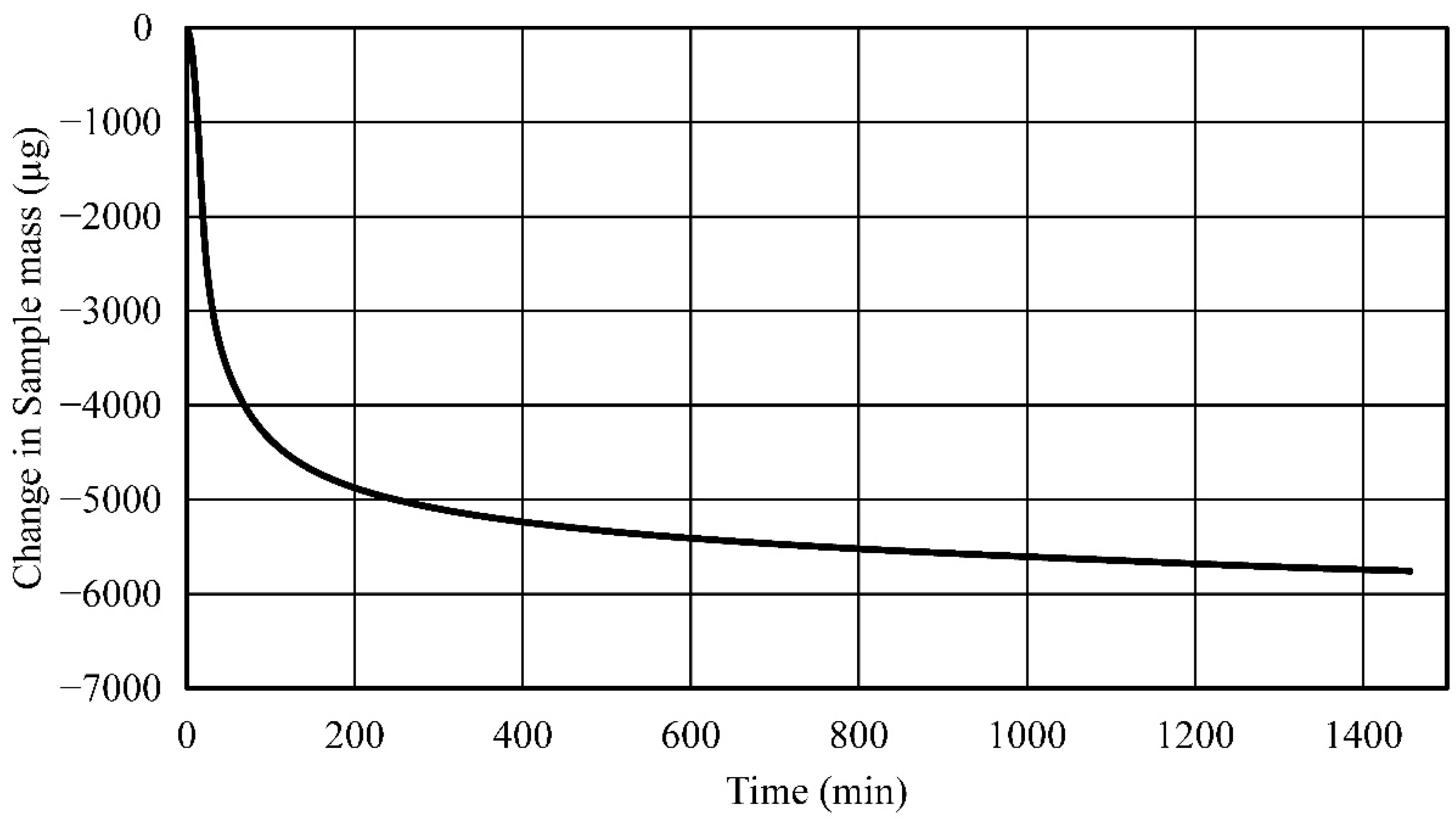
- Established process and process parameters to synthesize and 3D print carbonated polymer composites and studied effects of UV overexposure and post-heat treatment—it was observed that extended UV-exposure dose showed no effect on additional solvent removal. Post-heat treatment using thermogravimetric analysis of the neat polymer sample at elevated temperatures resulted in a 33% mass reduction, indicative of nearly complete water and solvent removal and curing.
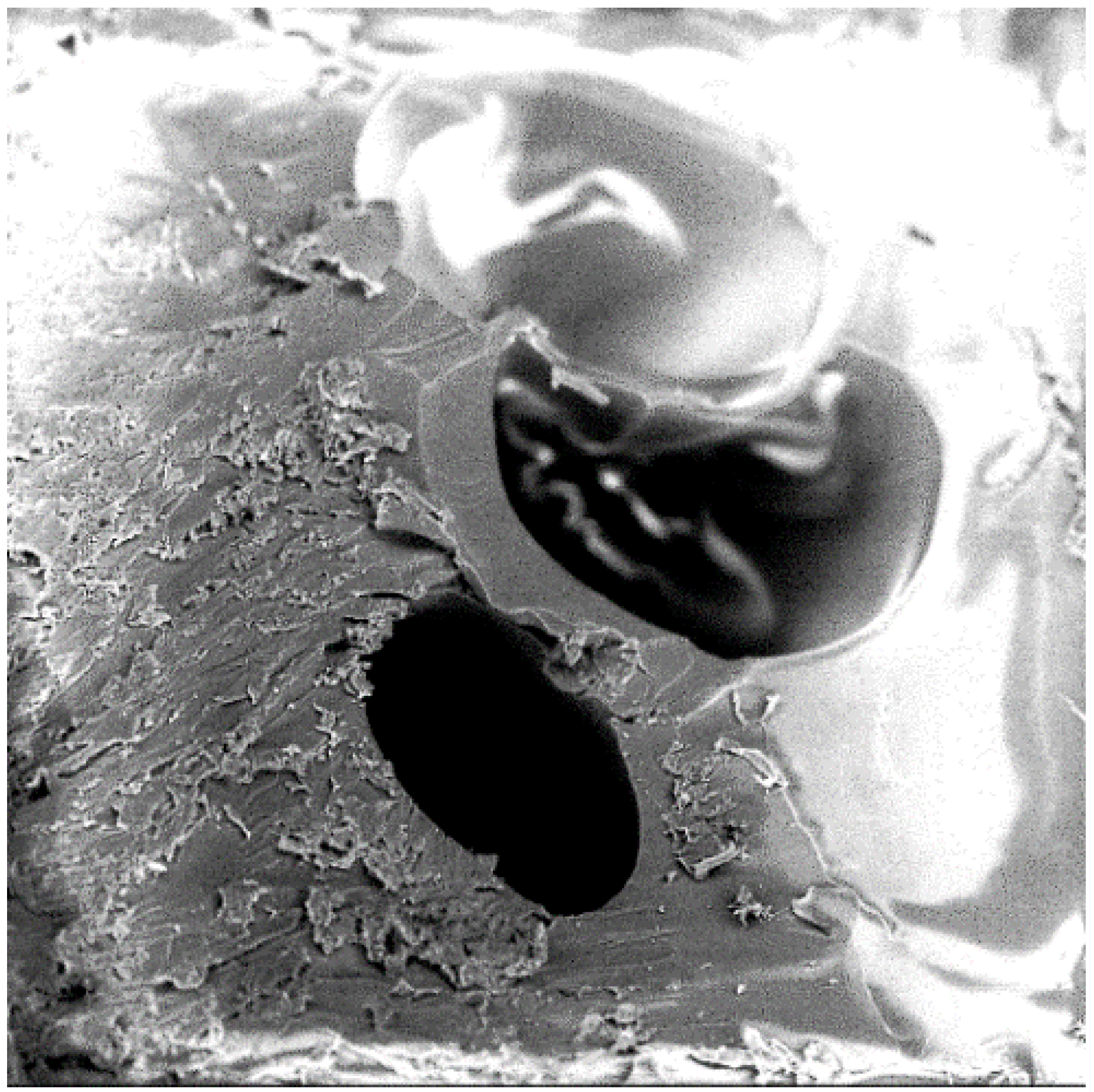
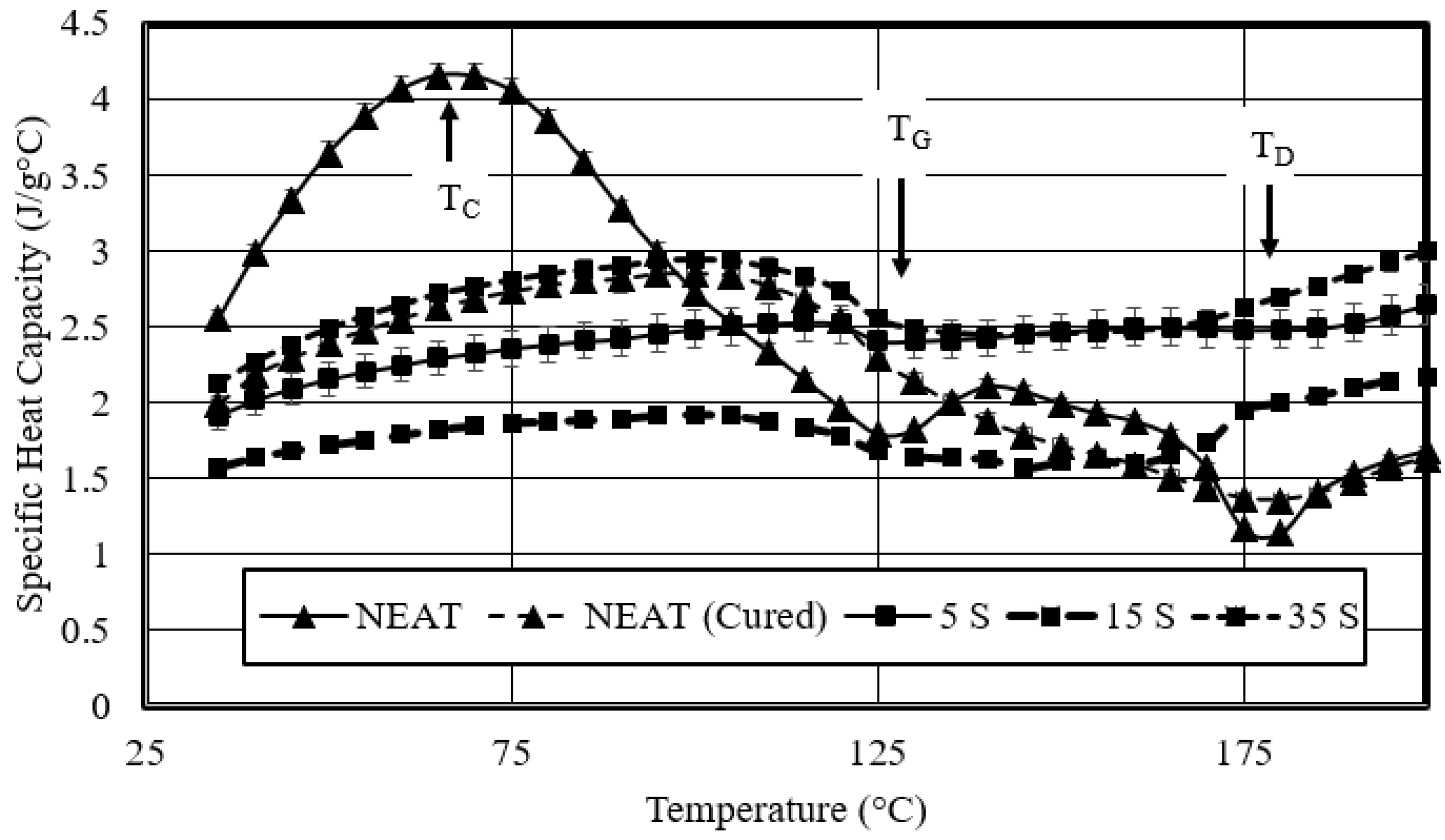
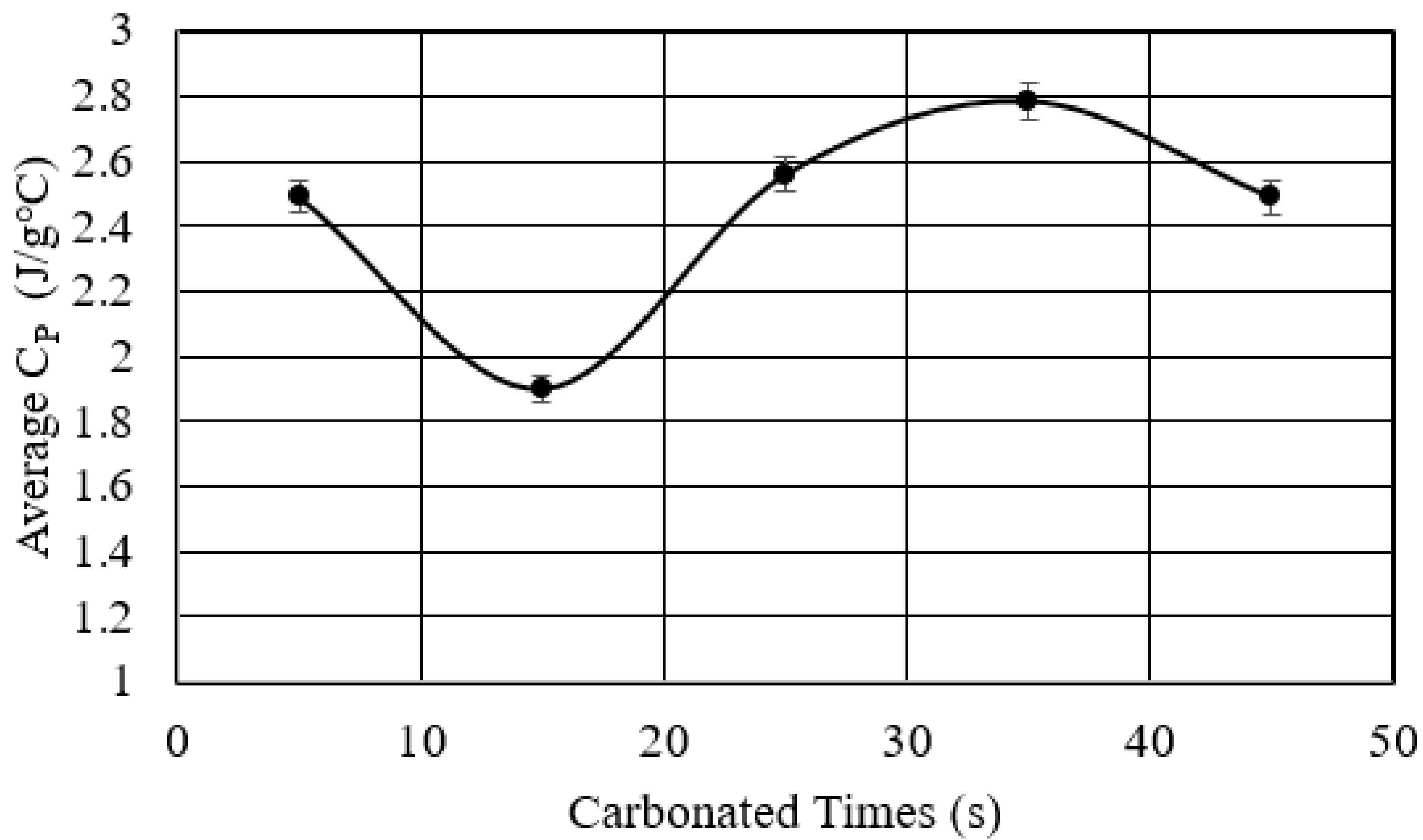
- Studied the effects of carbonation duration on polymer porosity and thermal profile behavior—an initial increase in polymer carbonation duration showed a 16% increase in porosity, more stable thermal profiles, and a 40% decrease in specific heat capacity.
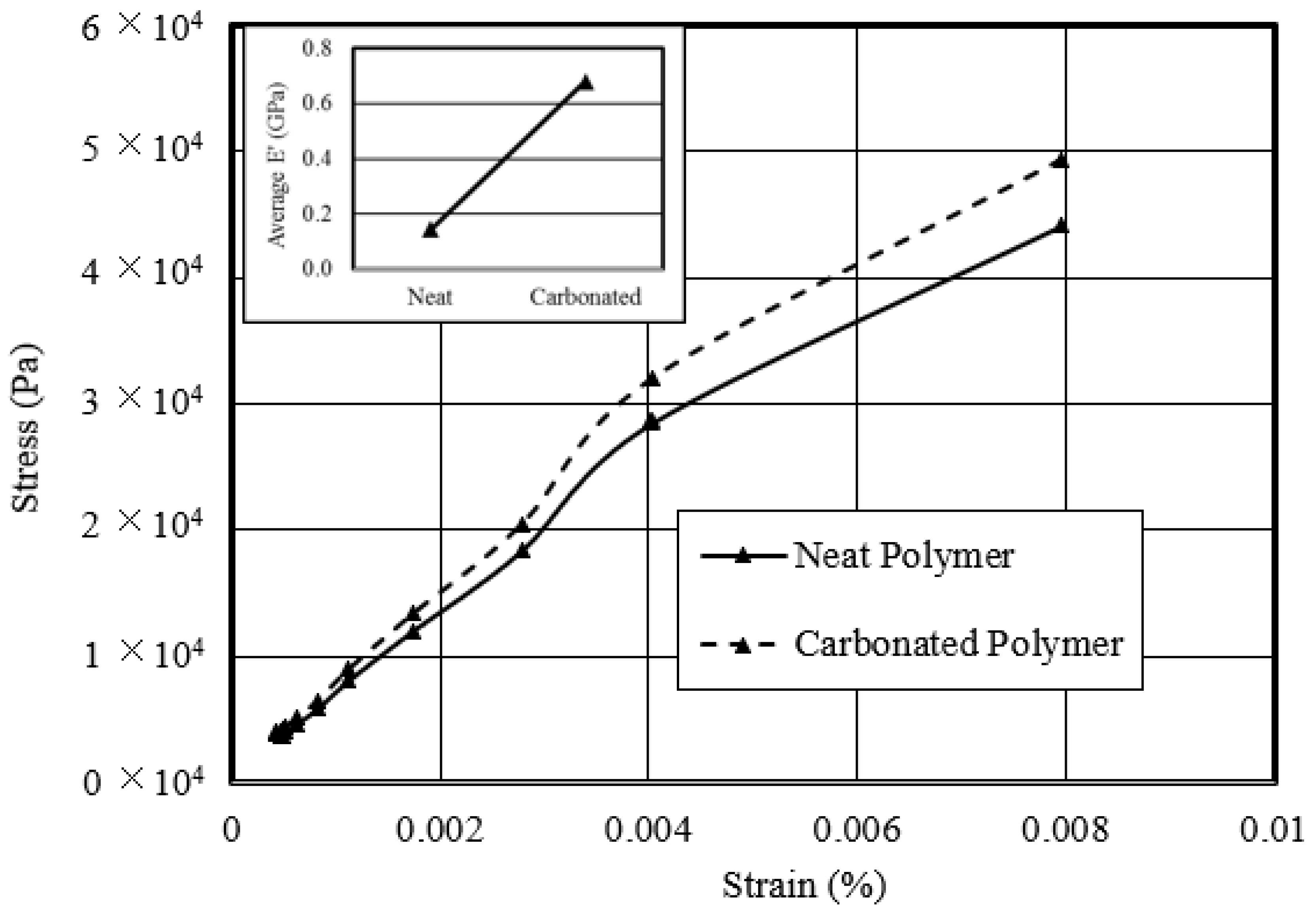
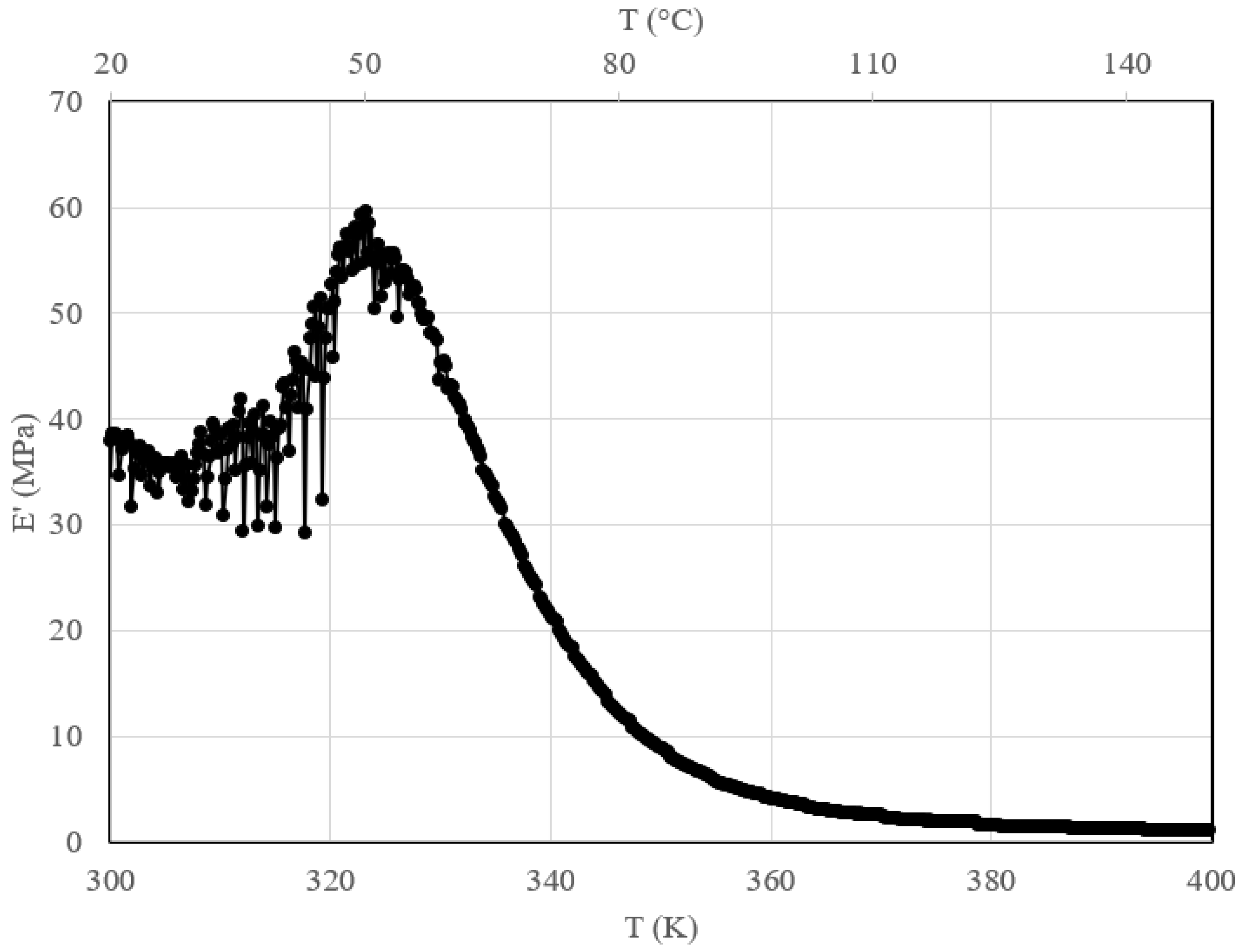
- Studied the response of the carbonated polymer to thermo-mechanical compressive tests—an optimum carbonated sample revealed a 70% increase in compressive strength over its neat counterpart. The temperature modulus-dependent behavior of the carbonated sample showed a peak modulus maximum at 50 °C of 60 MPa, after which it decreased exponentially with increased temperature.
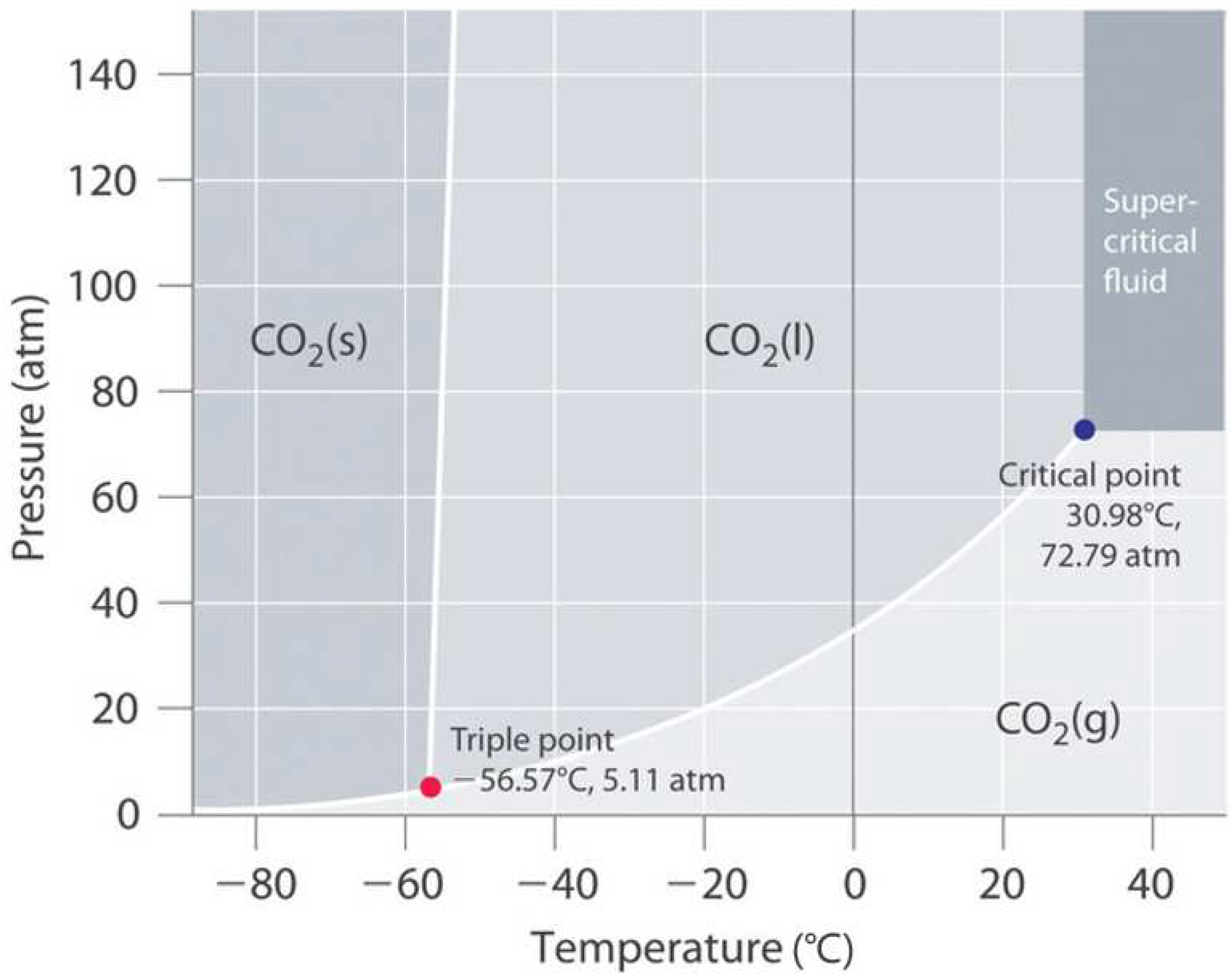
- Considered the application of the carbonated polymer composite given the results found—potential utility of the printed carbonated composite as a personal, portable, and on-demand CO2 phase transformation storage medium. The critical point to transform the gas into a supercritical fluid occurs at a minimum temperature and pressure of 30.98 °C and 72.79 atm (7.38 MPa), respectively. The thermo-mechanical results of this study determined that the manufactured composite is thermally and mechanically suitable for this application.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tjong, S.C. Structural and mechanical properties of polymer nanocomposites. Mater. Sci. Eng. R Rep. 2006, 53, 73–197. [Google Scholar] [CrossRef]
- Burela, R.G.; Kamineni, J.N.; Harursampath, D. 3D and 4D Printing of Polymer Nanocomposite Materials; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Dawan, F.; Jin, Y.; Goettert, J.; Ibekwe, S. High functionality of a polymer nanocomposite material for MEMS applications. Microsyst. Technol. 2008, 14, 1451–1459. [Google Scholar] [CrossRef]
- DeArmitt, C.; Rothon, R. Particulate Fillers, Selection, and Use in Polymer Composites. Polymers and Polymeric Composites: A Reference Series. In Encyclopedia of Polymers and Composites; Springer Nature Switzerland AG: Basel, Switzerland, 2016. [Google Scholar]
- Xu, Z.; Chen, Y.; Chen, X.; Zhang, J.; Huang, S.; Chen, A.; Fu, X.; Wu, F.; Zhang, P. Enhanced thermal conductivity and electrically insulating of polymer composites. J. Mater. Sci. 2021, 56, 4225–4238. [Google Scholar] [CrossRef]
- Mooti, A.; Costa, C.; Maceiras, A.; Pereira, N.; Tubio, C.; Vilas, J.; Besbes-Hentati, S.; Lanceros-Mendez, S. Magnetic and high-dielectric-constant nanoparticle polymer tri-composites for sensor applications. J. Mater. Sci. 2020, 55, 16234–16246. [Google Scholar] [CrossRef]
- Mehdikhani, M.; Gorbatikh, L.; Verpoest, I.; Lomov, S.V. Voids in fiber-reinforced polymer composites: A review on their formation, characteristics, and effects on mechanical performance. J. Compos. Mater. 2018, 53, 1579–1669. [Google Scholar] [CrossRef]
- Biner, S. The role of interfaces and matrix void nucleation mechanism on the ductile fracture process of discontinuous fibre-reinforced composites. J. Mater. Sci. 1994, 29, 2893–2902. [Google Scholar] [CrossRef]
- Jezzard, P.; Wiggins, C.J.; Carpenter, T.A.; Hall, L.D.; Barnes, J.A.; Jackson, P.; Clayden, N.J. Demonstration of nuclear magnetic resonance imaging for void detection in carbon-fibre reinforced polymer composites, and comparison with ultrasound methods. J. Mater. Sci. 1992, 27, 6365–6370. [Google Scholar] [CrossRef]
- Levi, G.; Kaplan, W. The influence of interfacial wetting and adhesion on the formation of voids at metal-ceramic interfaces. J. Mater. Sci. 2006, 41, 817–821. [Google Scholar] [CrossRef]
- Li, Y.; Li, Q.; Ma, H. The voids formation mechanisms and their effects on the mechanical properties of flax fiber reinforced epoxy composites. Compos. Part A Appl. Sci. Manuf. 2015, 72, 40–48. [Google Scholar] [CrossRef]
- Liebig, W.; Viets, C.; Schulte, K.; Fiedler, B. Influence of voids on the compressive failure behaviour of fibre-reinforced composites. Compos. Sci. Technol. 2015, 117, 225–233. [Google Scholar] [CrossRef]
- DeValve, C.; Pitchumani, R. Simulation of void formation in liquid composite molding processes. Compos. Part A Appl. Sci. Manuf. 2013, 51, 22–32. [Google Scholar] [CrossRef]
- Afendi, M.; Banks, W.M.; Kirkwood, D. Bubble free resin for infusion process. Compos. Part A Appl. Sci. Manuf. 2013, 36, 739–746. [Google Scholar] [CrossRef]
- Al-Ghussain, L. Global Warming: Review on Driving Forces and Mitigation. Environ. Prog. Sustain. Energy 2019, 38, 13–21. [Google Scholar] [CrossRef] [Green Version]
- GLOBAL CLIMATE CHANGE Vital Signs of the Planet. Available online: https://climate.nasa.gov (accessed on 15 March 2022).
- Topham, S.; Bazzanella, A.; Schiebahn, S.; Luhr, S.; Zhao, L.; Otto, A.; Dioxide, D.S.C. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH GmbH & Co. KgaA: Weinheim, Germany, 2014; pp. 1–43. [Google Scholar]
- Cooper, A.I. Porous Materials and Supercritical Fluids. Adv. Mater. 2003, 15, 1049–4059. [Google Scholar] [CrossRef]
- Song, B.; Qin, A.; Tang, B.Z. Syntheses, properties, and applications of CO2-based functional polymers. Cell Rep. Phys. Sci. 2022, 3, 100719. [Google Scholar] [CrossRef]
- Kim, J.; Kitagaki, R.; Choi, H. Pore Filling Effect of Forced Carbonation Reactions Using Carbon Dioxide Nanobubbles. Materials 2020, 13, 4343. [Google Scholar] [CrossRef]
- Puttonen, T.; Salmi, M.; Partanen, J. Mechanical properties and fracture characterization of additive manufacturing polyamide 12 after accelerated weathering. Polym. Test. 2021, 104, 107376. [Google Scholar] [CrossRef]
- Samsudin, S.S.; Ariff, Z.; Zakaria, Z.; Bakar, A. Development and characterization of epoxy syntactic foam filled with epoxy hollow spheres. Express Polym. Lett. 2011, 5, 653–660. [Google Scholar] [CrossRef]
- Zimmerman, D.L.; Jones, R.W. SEM Analysis of Polymeric Mechanical Failures in Polyetherimide. Int. J. Polym. Mater. Polym. Biomater. 1994, 23, 151–165. [Google Scholar] [CrossRef]
- Khosravani, M.R.; Berto, F.; Ayatollahi, M.R.; Reinicke, T. Fracture behavior of additively manufactured components: A review. Theor. Appl. Fract. Mech. 2020, 109, 102763. [Google Scholar] [CrossRef]
- Haugen, H.; Bertold, S.I. Characterization of morphology—3D porous structure. Charact. Polym. Biomater. 2017, 21–53. [Google Scholar]
- Wolok, E.; Pakaya, F. Experimental Investigation of Epoxy/Poly (amino amide)/Phthalic Anhydride: Mechanical Properties and Thermal Stability. Int. J. Adv. Sci. Technol. 2020, 29, 1011–1021. [Google Scholar] [CrossRef]
- ASTM D4892; Standard Test Method for Density of Solid Pitch (Helium Pycnometer Method). American Society for Testing and Materials: West Conshohocken, PA, USA, 2019.
- E1269–11; Standard Test Method for Determining Specific Heat Capacity by Differential Scanning Calorimetry. American Standards for Testing Materials: West Conshohocken, PA, USA, 2011.
- Richardson, M. Thermal Analysis of Polymers using Quantitative Differential Scanning Calorimetry. Polym. Test. 1984, 4, 101–115. [Google Scholar] [CrossRef]
- Bilyeu, B.; Brostow, W.; Menard, K.P. Epoxy Thermosets and Their Applications. II. Thermal Analysis. J. Mater. Educ. 2000, 22, 107–129. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Thickness (µm) | UV Dose/Time |
|---|---|---|
| Carbonation duration | - | 0 to 45 s |
| Printed base layer | 200 | 86 mJcm−2 |
| Printed sub-layers | 50 | 3.8 mJcm−2 |
| IPA Rinse | - | 2 min spray |
| Air dry and storage | - | 5 min + 24 h dry dark storage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawan, F.; Givens, M.; Williams, L.; Mensah, P. Carbonated 3D-Printable Polymer Composite for Thermo-Mechanically Stable Applications. J. Manuf. Mater. Process. 2022, 6, 66. https://doi.org/10.3390/jmmp6030066
Dawan F, Givens M, Williams L, Mensah P. Carbonated 3D-Printable Polymer Composite for Thermo-Mechanically Stable Applications. Journal of Manufacturing and Materials Processing. 2022; 6(3):66. https://doi.org/10.3390/jmmp6030066
Chicago/Turabian StyleDawan, Fareed, Melvin Givens, Lakeira Williams, and Patrick Mensah. 2022. "Carbonated 3D-Printable Polymer Composite for Thermo-Mechanically Stable Applications" Journal of Manufacturing and Materials Processing 6, no. 3: 66. https://doi.org/10.3390/jmmp6030066
APA StyleDawan, F., Givens, M., Williams, L., & Mensah, P. (2022). Carbonated 3D-Printable Polymer Composite for Thermo-Mechanically Stable Applications. Journal of Manufacturing and Materials Processing, 6(3), 66. https://doi.org/10.3390/jmmp6030066