1. Introduction
Abdominal adipose tissue is considered to be particularly pathogenic in nature with increasing obesity [
1]. Adipose tissue is an endocrine organ that secretes numerous bioactive peptides often referred to as adipocytokines, which have been proposed to participate in the development of hypertension [
2]. From a basic physiological perspective, adipose tissue growth is tightly coupled to angiogenesis and microcirculation development. Tumor necrosis factor-alpha (TNF-α) is known to regulate angiotensinogen (AGT) in hepatocytes, as the AGT promoter contains a cytokine-inducible enhancer known as the acute phase response element [
3].
Pro-inflammatory adipocytokine secretion from adipose tissue is coordinated through the activation of the nuclear factor-kappa B (NF-κB) transcription factor [
4], which regulates the transcription of genes involved in inflammatory responses, cell growth control, and apoptosis. Thus, additional pathways in the development of hypertension may emanate from NF-κB activation. In resting cells, NF-κB is bound to I kappa B (IκB) inhibitors, which prevent NF-κB from entering the nucleus.
Extracellular stimuli such as pro-inflammatory cytokines, chemokines, and lipopolysaccharides (LPS) activate a set of proteins termed IκB kinases (IKKs), which phosphorylate IκB leading to an altered conformation of IκB, which releases NF-κB to enter the nucleus and activates transcription of target genes [
5]. Lipopolysaccharides increase the secretion of cytokines such as TNF-α via NF-κB activation [
6]. TNF-α also induces the transcription of AGT via NF-κB [
7]. Moreover, there is accumulating evidence that indicates angiotensin (ANG) II is capable of inducing an inflammatory response in the cardiac tissue through the activation of NF-κB [
8].
However, the study of NF-κB has, to some extent, been hampered by the lack of selective and specific inhibitory compounds. Our study, therefore, investigated the role of a cell-permeable peptide, SN50, as an NF-κB inhibitor. This peptide consists in the nuclear localization sequence of NF-κB subunit (p50) fused with the hydrophobic region of the signal sequence of Kaposi’s fibroblast growth factor. Lin et al. [
9] demonstrated that SN50 is capable of inhibiting the nuclear import of NF-κB in human monocytic cells and murine endothelial cells stimulated with LPS and TNF-α.
We hypothesized that the activation of NF-κB, and the subsequent secretion and expression of TNF-α, and AGT from human adipose tissue may play an important role in the development of arterial hypertension in obese subjects. The results may enable us to understand the role of various pro-inflammatory agents upregulated by activated NF-κB in the development of arterial hypertension. Our study was aimed to ascertain the effect of the NF-κB inhibitor, SN50, on reducing TNF-α and AGT secretion and expression in mediating the anti-inflammatory effect through its effects on the NF-κB inflammatory pathway in human adipose tissue.
2. Materials and Methods
2.1. Subjects and Adipose Tissue
Abdominal subcutaneous adipose tissue (AbdScAT) samples were obtained from 10 overweight (mean age: 30.71 ± 9.69; and a mean BMI: 27.92 ± 0.66) and 10 obese subjects with and without hypertension (mean age: 41.6 ± 12.62; and a mean BMI: 36.245 ± 6.95). They were undergoing abdominal adipose tissue liposuction for cosmetic reasons. No subject on any regular medication was taken. Subjects with chronic debilitating diseases were excluded from the study. All subjects gave their informed consent. Personal information and anthropometric measurements were taken from each subject. The previous history of diabetes, hypertension, and/or any risk factors were mentioned. All subjects fasted overnight before surgery, and only normal saline was administered intravenously until the tissue liposuction was taken. The study was carried out at the Chair for Biomarkers of Chronic Diseases and Obesity Research Center, King Saud University, Riyadh, KSA. Ethical approval was granted by the ethics committee of the King Khalid University Hospital, College of Medicine, King Saud University, Riyadh, KSA.
2.2. Isolation and Culture of Mature Adipocytes
Adipose tissue liposuction samples were digested for 30 min at 37 °C in Hank’s balanced salt solution (HBSS) containing 2 mg/mL collagenase under intermittent shaking as described by [
10]. The mixture was then centrifuged at 2000 rpm for 5 min at room temperature. After centrifugation, the tubes were tilted, allowing the densely packed adipocytes layer (cake of fat cell) to flow gently into a clean falcon tube. Mature fat cells were then washed by pouring in 10–15 mL of Dulbecco’s Modified Eagle Medium/Nutrient Mixture
(DMEM) F-12 phenol-red free (1 mL containing 15 mmol/L glucose, supplemented with 100 U/mL of penicillin and 100 µg/mL streptomycin). Fat cells were centrifuged at 1000 rpm for 1 min. Once the adipocytes had been washed, the liquid was removed from beneath and the cake was collected into a new falcon tube. Following isolation of these cells, cells (0.2 mL, 100,000 adipocytes) were plated in 6-well tissue culture plates with a culture medium (1 mL containing 15 mmol/L of glucose and supplemented with 100 U/mL of penicillin, 100 µg/mL streptomycin, and 100 µg/mL transferrin). The cells were preincubated with SN50 and SN50M peptides (50 µg/mL) before the initiation of stimulation to enable efficient membrane translocation at 37 °C in 95% air and 5% CO
2 for 2 h. After preincubation, the cells were treated with 10 ng/mL LPS for 03, 12, 24, and 48 h before the experiments. Initial experiments were undertaken to evaluate the optimum concentration of NF-κB inhibitor (concentration range: 1, 10, 50, and 100 µg/mL SN50) and LPS (concentration range: 1, 10, and 100 ng/mL) without causing cellular death; these were deemed as 50 µg/mL SN50 and 10 ng/mL LPS. Following treatment, conditioned media and adipocytes were separated by centrifugation at 1000 rpm for 1 min. After centrifugation, the infranatant was separated and stored at −80 °C until adipokine measurement, whereas fat cells were used for protein expression (NF-kB p65 activity assay and western blot) and gene expression of TNF-α and ANG.
2.3. NF-κB p65 Activity Assay
NF-κB p65 activity was assessed using the transcription factor assay kit according to the manufacturer’s instructions (TransAM NF-κB p65 kit, Active Motif, Rixenart, Belgium; cat. no. 40096), as described by [
11]. In brief, 20 µg of total protein were used to detect NF-κB p65 activity. Jurkat cell nuclear extract (2.5 µg) was used as a positive control for NF-κB p65 activation. This extract is optimized to give a strong signal when used at 2.5 µg/well. Wild-type consensus oligonucleotides were used in the assay as a competitor for NF-κB binding to monitor the specificity of the assay. Twenty picomoles per well of used oligonucleotides was enough to prevent NF-κB binding to the probe immobilized on the plate. Conversely, the mutated consensus oligonucleotides were used as an noncompetitor for NF-κB binding.
2.4. Isolation and Protein Quantification
The total protein amount was quantified using the Bradford DC (detergent compatible) protein assay kit (Bio-Rad, USA, cat. no. 500-0111) as described by Bradford [
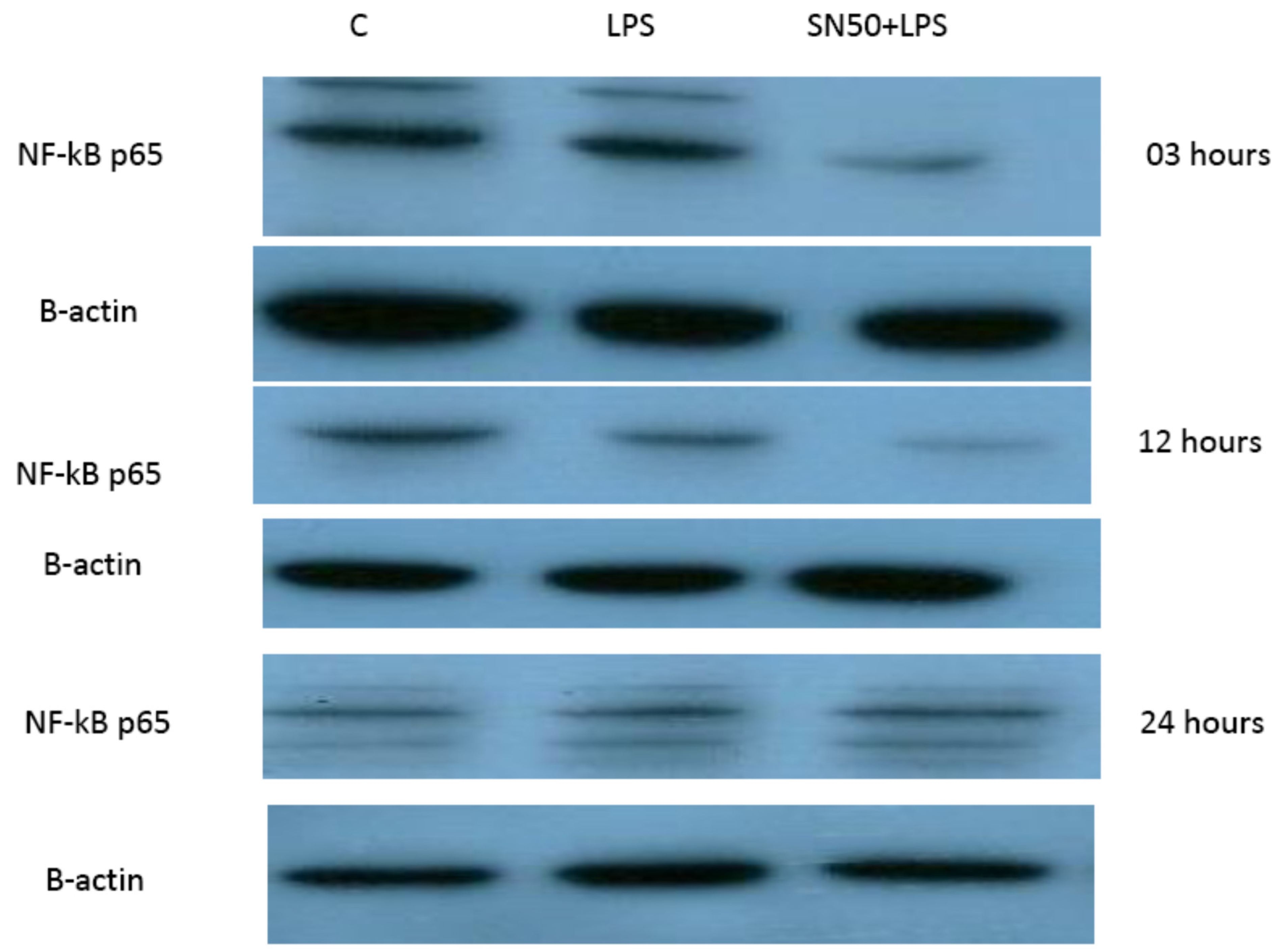
12]. Equal amounts of protein from each sample (20 µg) and pertained markers were heated for 5 min at 95 °C in a loading sample buffer, loaded, and separated by 12% sodium dodecyl sulphate-polyacrylamide (SDS-PAGE) running gel and 4% SDS-PAGE stacking gel. Gels were run for 2 h at 120 volts and then blotted and transferred onto polyvinylidene fluoride (PVDF) membrane for 25 min at 15 volts using a liquid transfer system. After transfer, the membrane was washed with 1X tris-buffered solution (TBS) for 5 min. The membrane was blocked for 1 h at room temperature in a blocking buffer containing TBS with 0.1% Tween20, 5% nonfat dried milk, and 1% bovine serum albumin (BSA). The membrane was then incubated using gentle agitation overnight at 4 °C with antibodies against the following: polyclonal rabbit IgG NF-kB p65 (1:1000) (Cell Signaling Technology), polyclonal rabbit IgG angiotensinogen (1:200) (Phoenix Pharmaceuticals Industries, Riyadh, Saudi Arabia), and polyclonal goat IgG TNF-α (1:1000) (R&D Systems, Minneapolis, MN, USA). After incubation, the membrane was then washed three times with TBS/T for 5 min each wash. The membrane was then incubated with the appropriate horse radish peroxidase (HRP)-conjugated secondary antibody with gentle agitation for one hour and washed five times for 5 min with TBS/T. The membrane was incubated with chemiluminescent development (luminol and peroxide substrate) with gentle agitation for 1 min in a dark room, and the membrane was drained, wrapped in plastic wrap, and exposed on the X-ray image.
2.5. Isolation and Purification of Total RNA
Cells from 6 wells were extracted with 200 μL of RNA (for RNA stabilization). Total RNA was isolated using the RNeasy mini kit according to the manufacturer’s instructions (Qiagen, GmBH, Hilden, Germany). RNA concentration and purity were performed using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.6. cDNA Synthesis
The reverse transcription step was conducted on 200 ng RNA using the QuantiTect reverse transcription kit (Qiagen, Valencia, CA, USA) as described by Heller et al. [
13]. In brief, purified RNA samples (200 ng/µL) were incubated with 2 µL of genomic DNA wipeout buffer at 42 °C for 2 min to effectively remove contaminating genomic DNA. After genomic DNA elimination, the RNA samples were reverse-transcribed using a master mix prepared from Quantiscript reverse transcriptase (1 µL), RT primer mix (1 µL), and Quantiscript RT buffer (4 µL) at 42 °C for 15 min.
2.7. Quantitative Real-Time PCR
All the experiments were performed in 96 well plates with the CFX96 Real-Time PCR detection system (Bio-Rad, Hercules, CA, USA) using a premade TaqMan probe (for AGT: Hs01586213_m1 and TNF-α Hs00174128_m1). Real-time relative expression experiments were performed according to the manufacturer’s instructions. In brief, 1 µL of cDNA was used in a final PCR volume of 20 µL, containing 10 µL of the TaqMan Master Mix (AB Applied Biosystems, Warrington, UK), 8 µL of RNA-free water, and 1 µL of the TaqMan probe. Polymerase chain reaction cycles were as follows: 10 min at 42 °C followed by 40 cycles for 15 s at 95 °C and 1 min at 60 °C. All reactions were multiplexed with the housekeeping gene human 18S ribosomal RNA (AB Applied Biosystems, Warrington, UK), which was used as a reference, enabling data to be expressed as delta cycle threshold (∆CT) values (where ∆CT = CT18s − CTgene of interest). Quantification of target mRNA was carried out by comparison of the number of cycles required reaching the reference and target threshold values (ΔΔCT method). Reactions were performed in triplicate for each sample. All statistics were performed at the ∆CT stage to exclude potential bias due to the averaging of data transformed through a 2-∆∆CT equation.
2.8. Statistical Analysis
Data were analyzed using the Statistical Package for Social Sciences (SPSS) for Windows (version 16.0 SPSS Inc., Chicago, IL, USA). Data are expressed as means ± standard errors (SEs). An independent sample t-test was done for delta CT (∆CT) among untreated and various treatment groups. p < 0.05 was considered as a significant value.
4. Discussion
Despite the high prevalence of obesity and hypertension in Saudi Arabia, to date, no study has examined the relationships between pro-inflammatory adipokines and blood pressure in the obese, hypertensive phenotype as well as the role of signaling pathways in the development of hypertension in human adipose tissue. As obesity and hypertension are increasingly considered to develop through subclinical chronic inflammation, the role of the pro-inflammatory adipokines becomes increasingly important to understand [
14]. Our study, focused on the NF-κB pathway as one signaling pathway contributing to the upregulation of pro-inflammatory adipokines such as TNF-α as well as RAS components like AGT in human adipose tissue, whose secretions in adipose tissue are coordinated through NF-κB activation. Harvested and isolated abdominal subcutaneous adipose tissue was used for this study.
Adipose tissue plays an important role in the secretion of certain pro-inflammatory adipokines and activation of these adipokines is coordinated through the NF-κB-dependent pathway [
4,
15,
16,
17]. The present study examined, in vitro, the effect of an NF-κB blocker (SN50) on TNF-α secretion as well as mRNA, TNF-α protein, and AGT expression in primary human adipocytes, which were isolated from overweight subjects and obese subjects with and without hypertension and treated with 10 ng/mL LPS as a potent stimulant pathogen at different concentrations and time points.
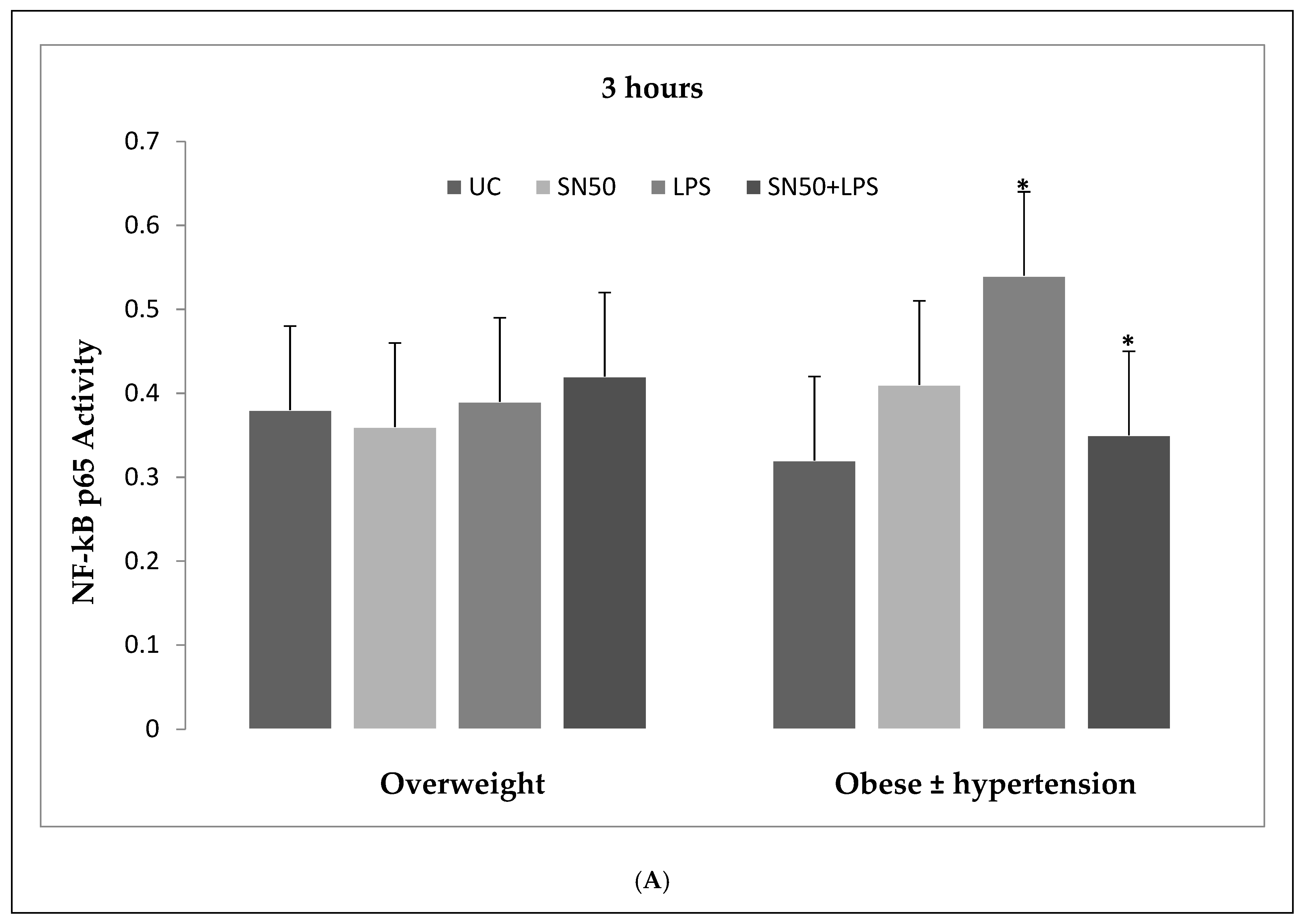
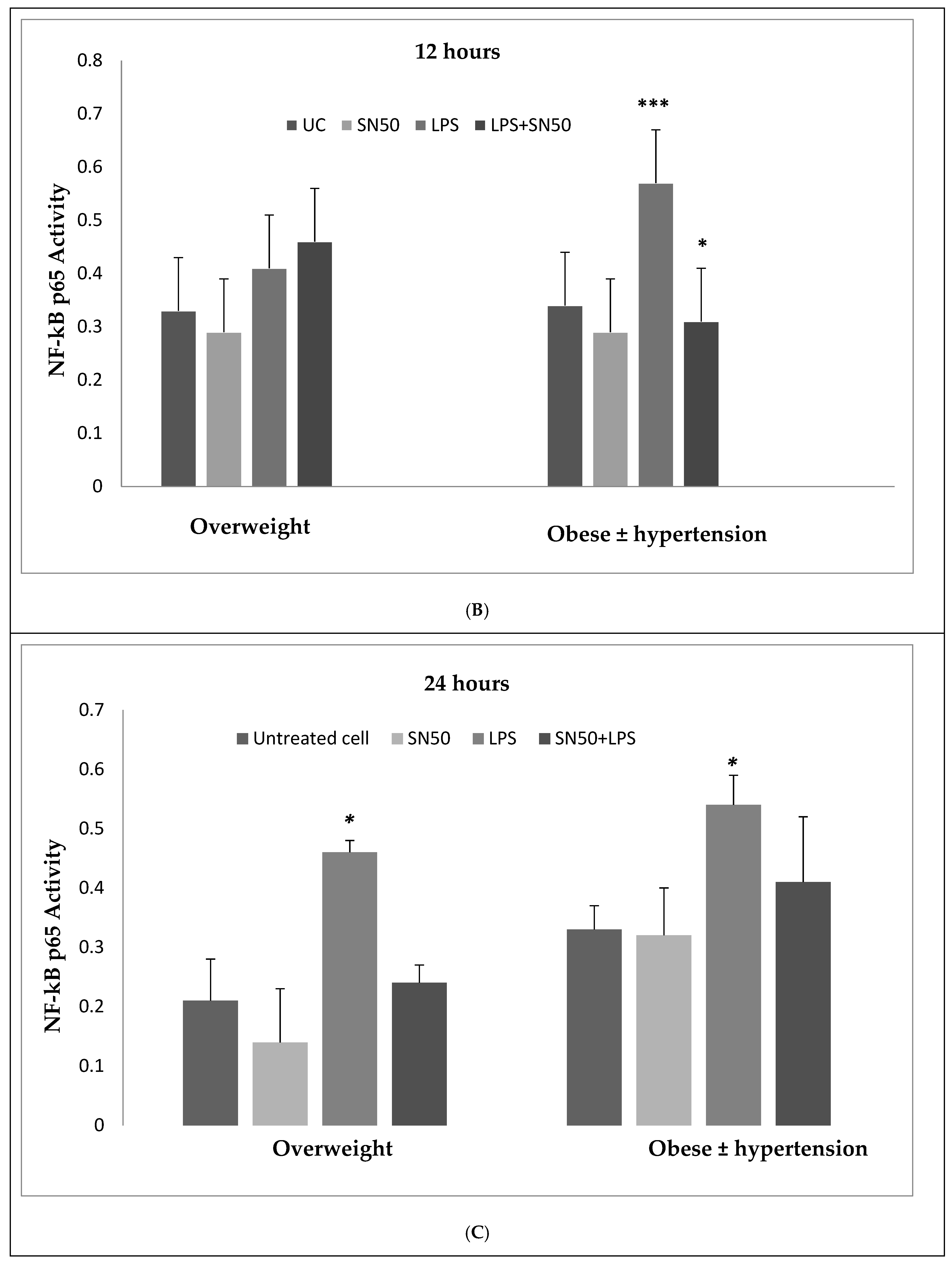
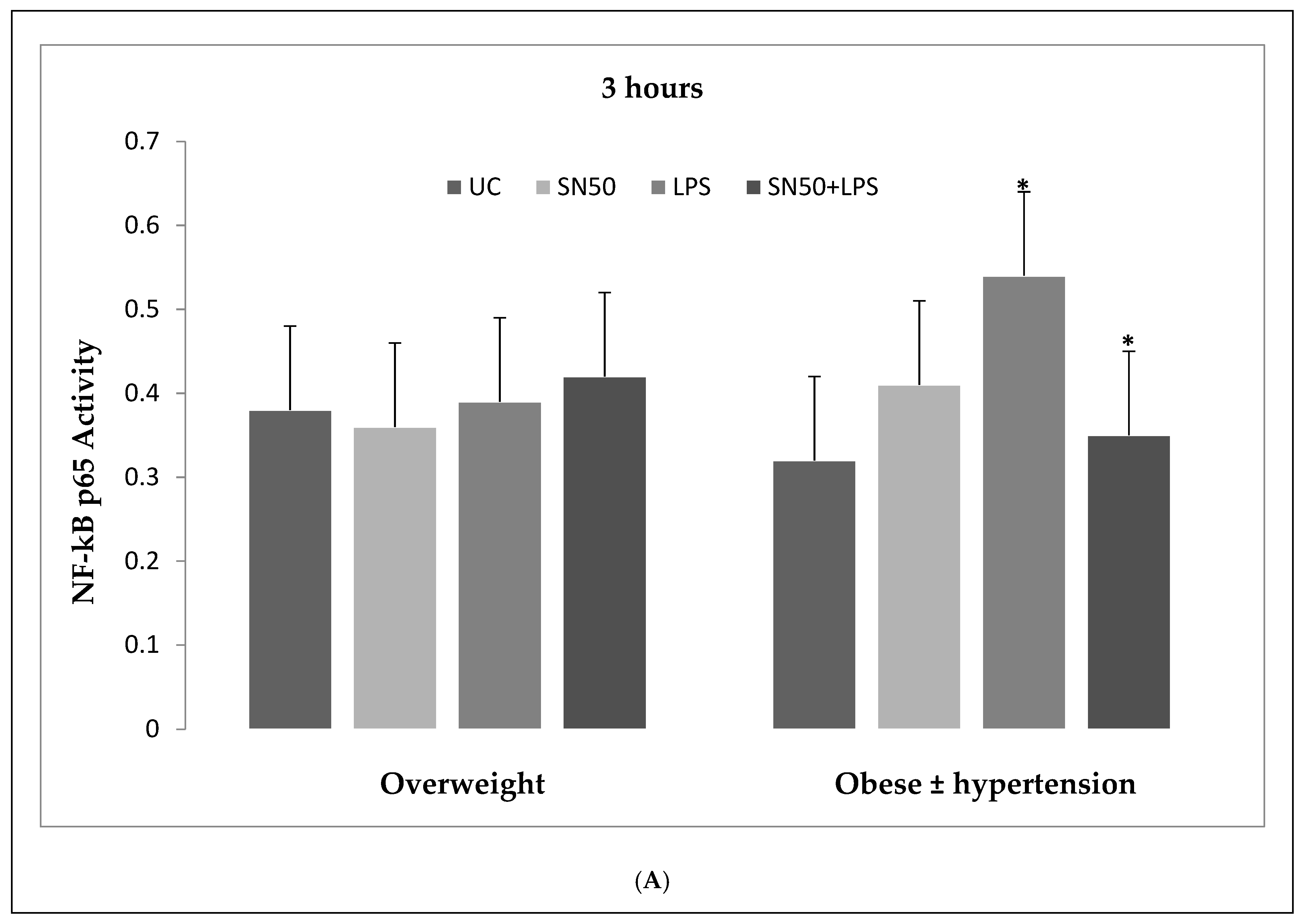
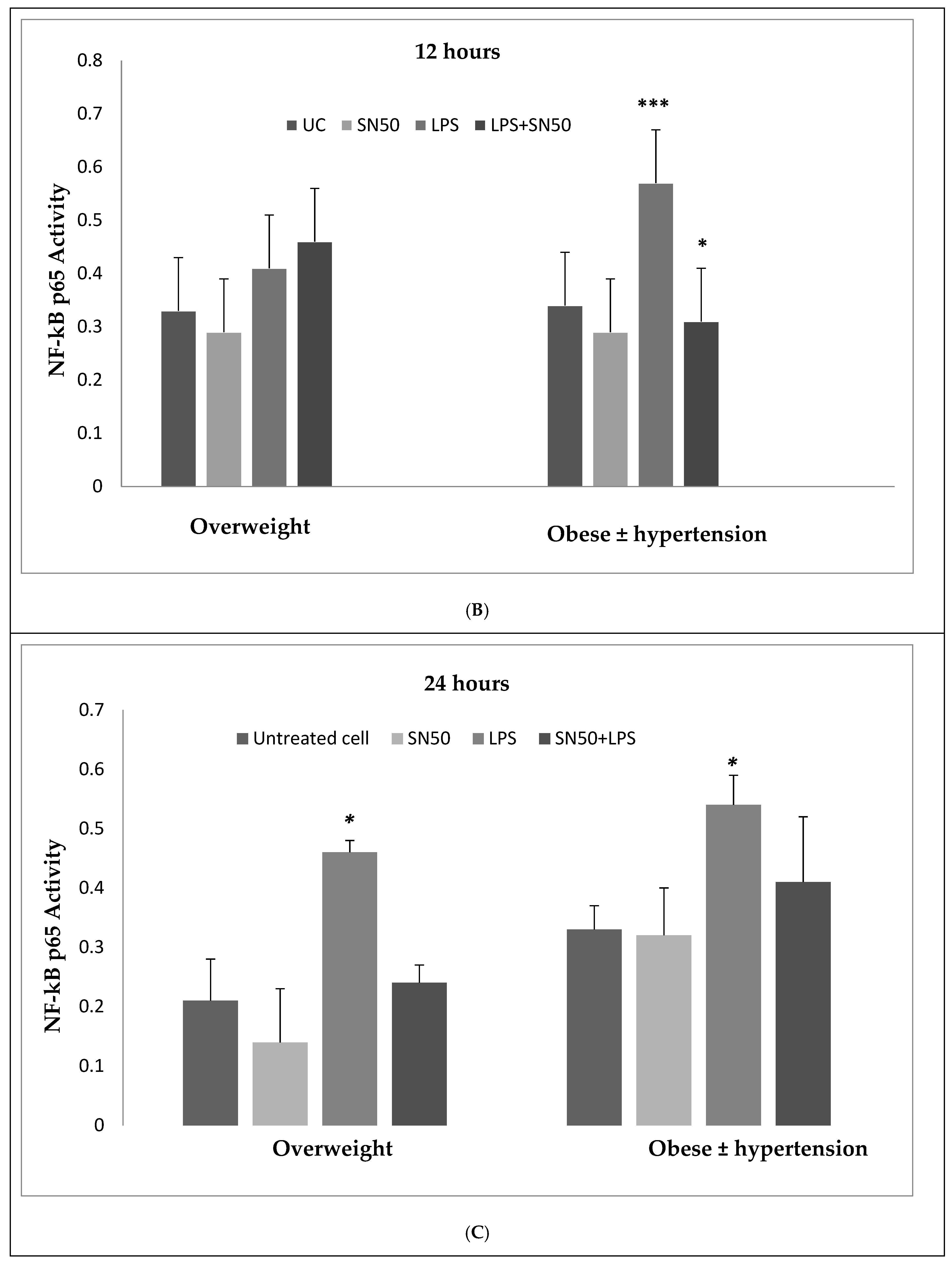
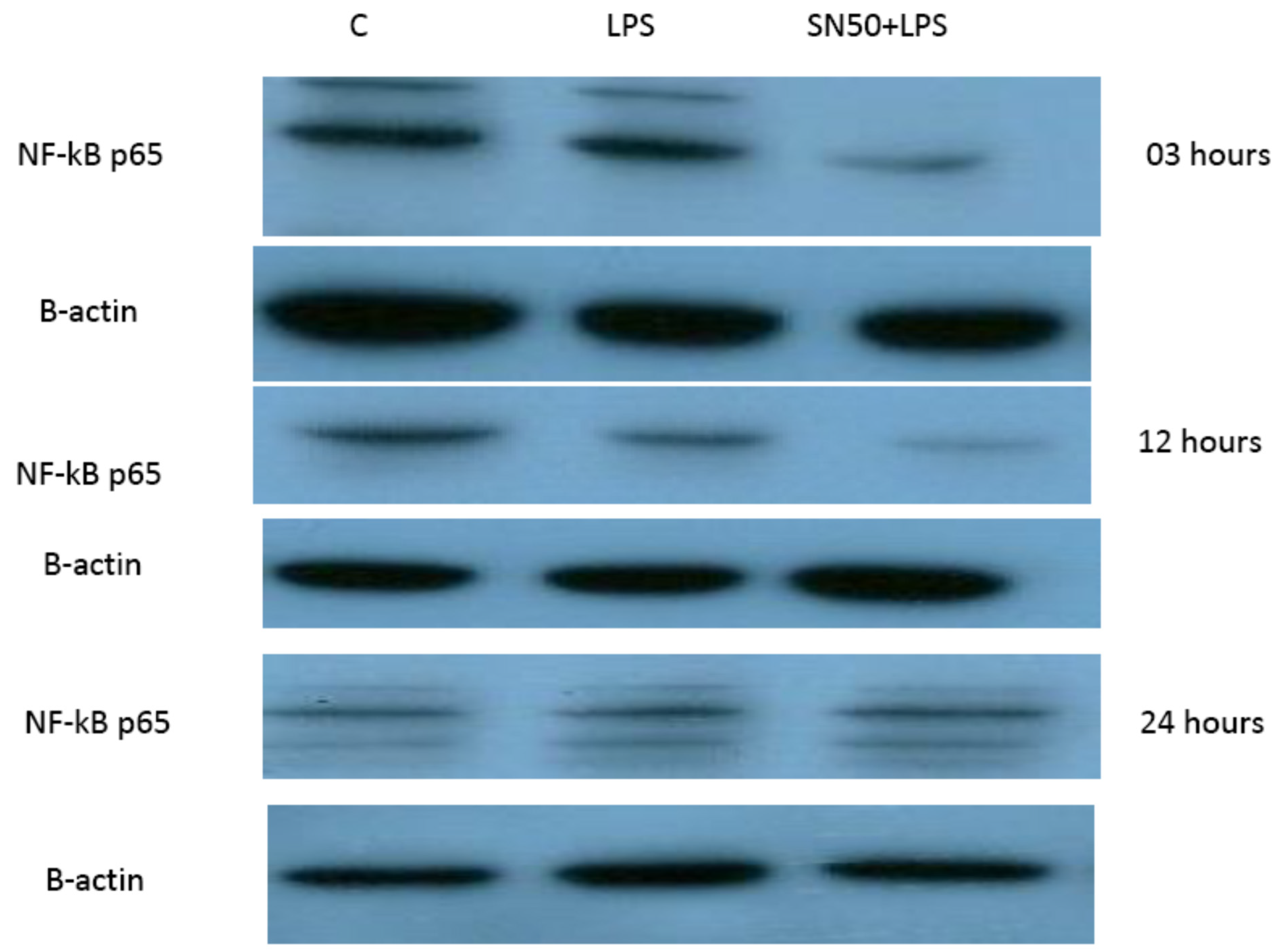
Our findings demonstrated that LPS significantly stimulates NF-κB activation in a concentration higher or equivalent to Jurkat cells (positive control cells) in both groups. Lipopolysaccharide also increase NF-κB p65 protein levels compared with those in control cells at 3 and 12 h and did not have an effect at 24 h. In contrast, coincubation with LPS and SN50 resulted in a marked reduction in NF-κB p65 levels as compared with those treated only with LPS. However, SN50 alone did not affect LPS through NF-kB activation and expression. Our findings suggest that SN50 at least in part suppresses NF-κB-mediated inflammatory pathways in adipose tissue. Another interesting finding is the relationship between NF-κB activation and the degree of adiposity. Our study observed a strong relationship between body mass index (BMI) and NF-κB p65, which remained significant independent of age.
Liposaccharide is a well-preserved component of the external part of the Gram-negative bacterial cell wall [
18]. This molecule is recognized by the innate immune system via toll-like receptors (TLRs)—a class of proteins that play a key role in the innate immune system, which recognize antigens including LPS, on monocyte/macrophage activation. Activation of TLRs (particularly TLR-4) leads to translocation of NF-κB into the nucleus to initiate gene expression of cytokines like IL-1, IL-6, and TNF-α [
18,
19,
20].
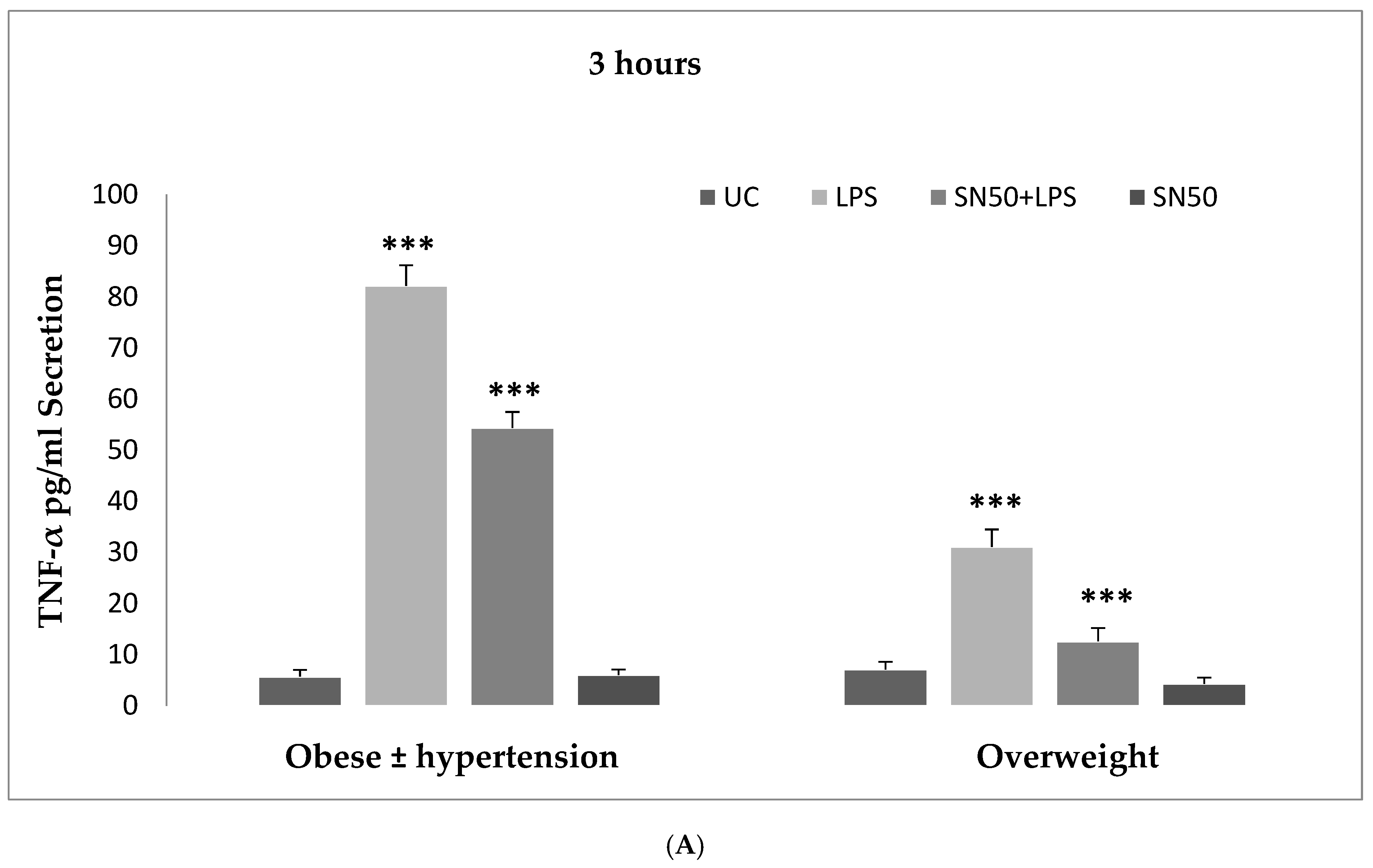
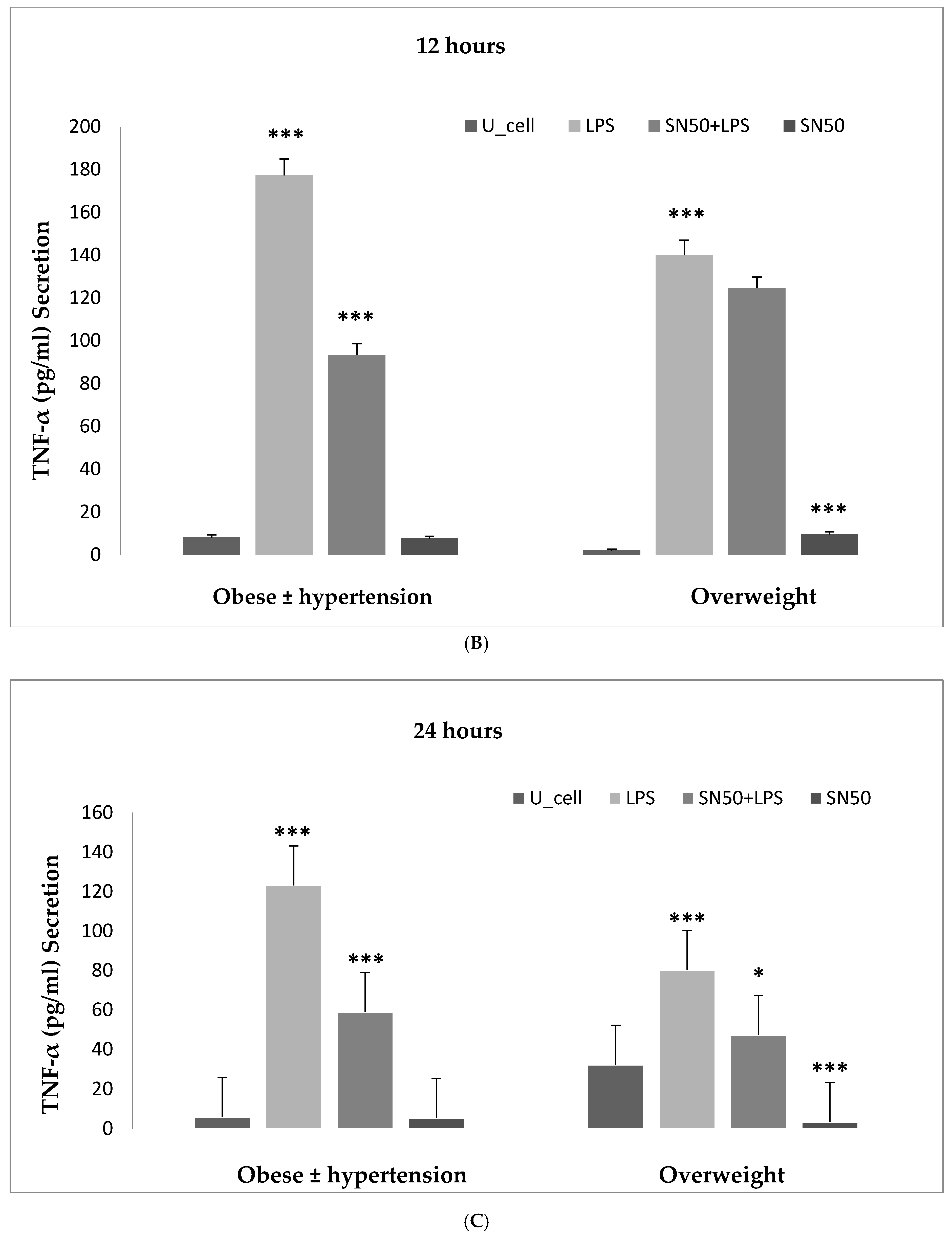
Numerous studies have shown that the endotoxin LPS has a potent inflammatory stimulant on cytokine secretion through NF-κB activation [
4,
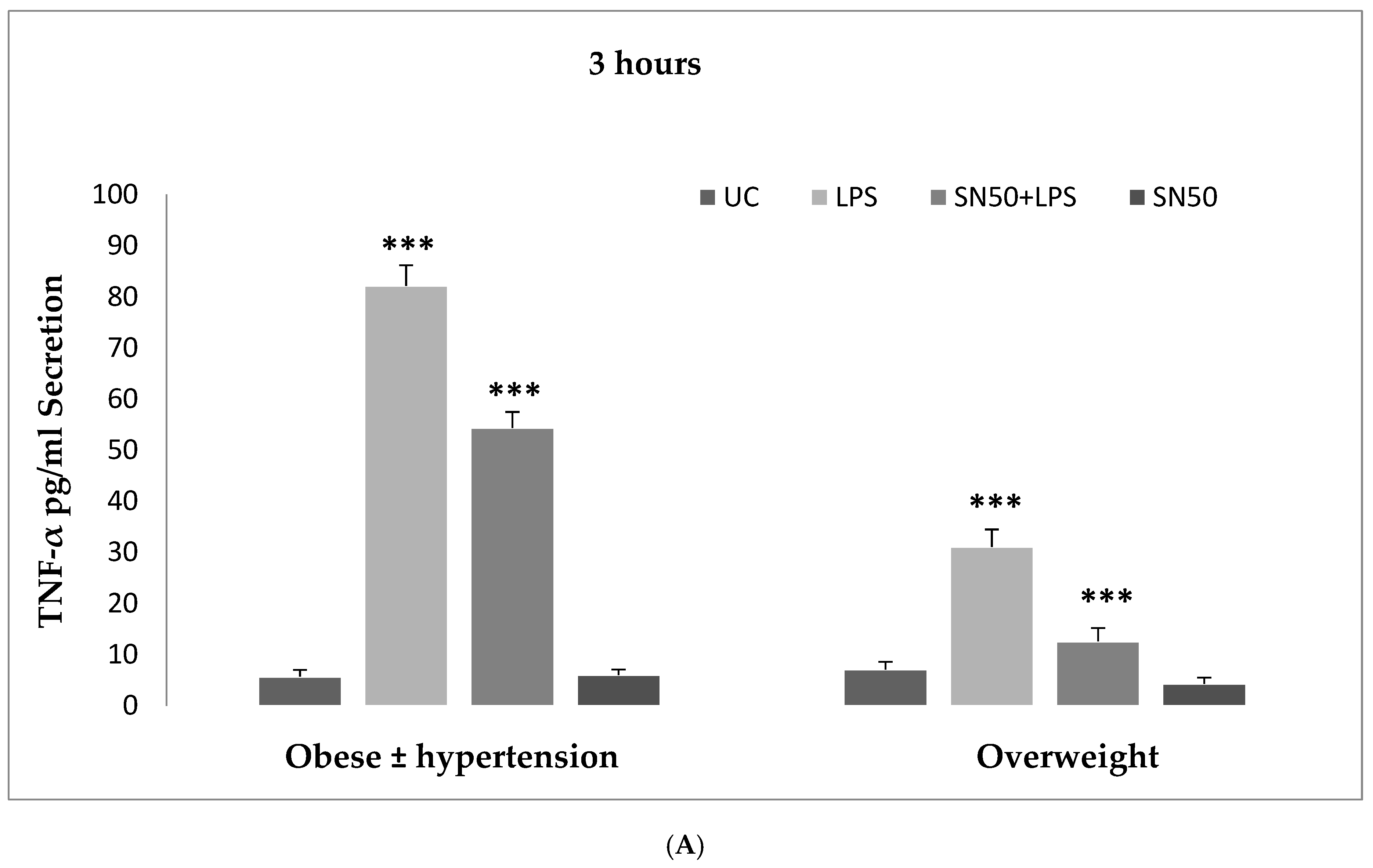
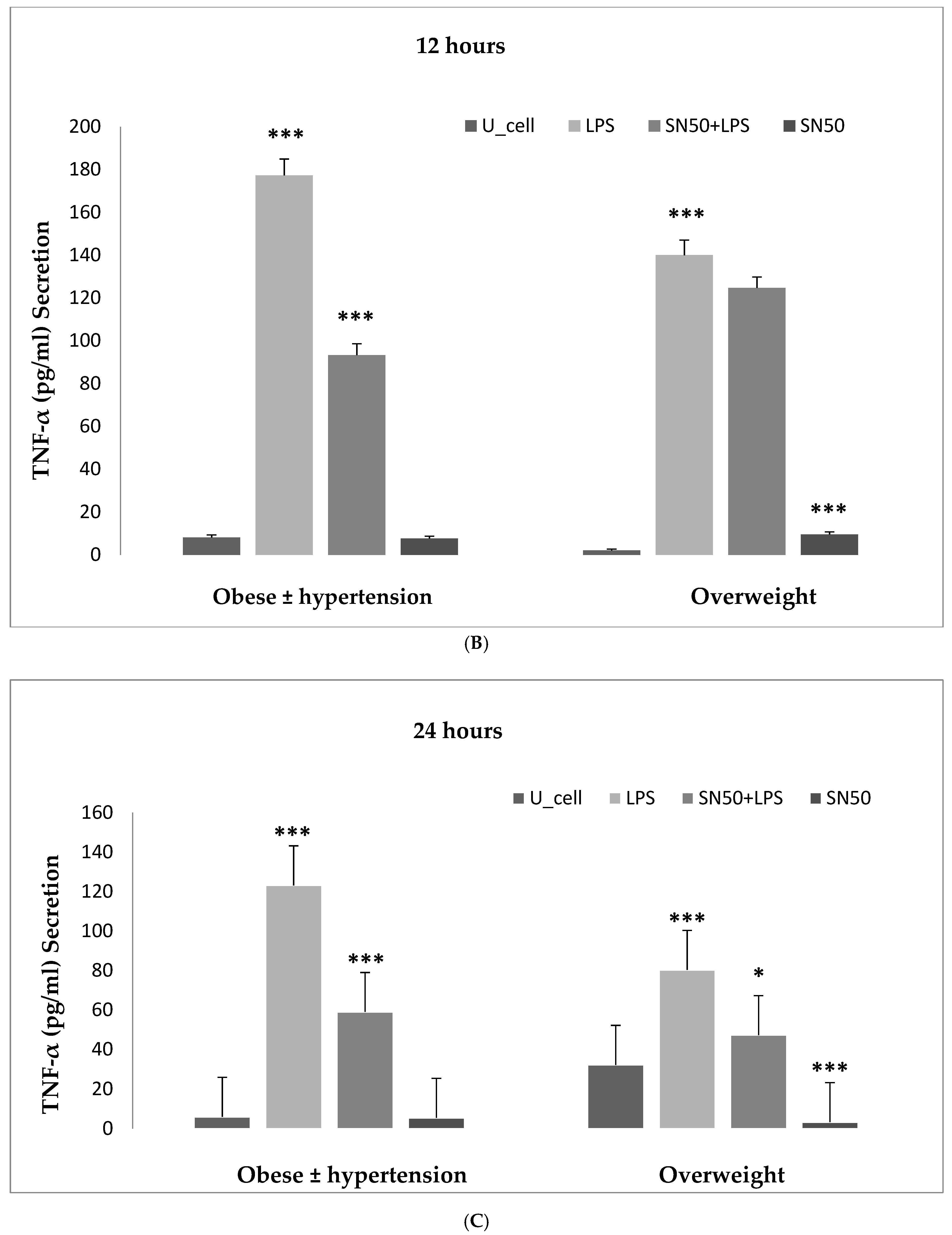
6]. Hence, our study addressed the principal activation regulatory pathways of the secretion of TNF-α by LPS via NF-κB activation. Treatment of adipocytes with 10 ng/mL LPS caused a significant increase in TNF-α secretion, whereas, a significant decrease occurred in response to the presence of SN50 as compared with those cells treated with LPS at 3, 12, and 24 h. The maximum inhibitory action of SN50 on NF-κB activation was obtained with a concentration of 50μg/mL without causing cell death, which was observed at 12 h after treatment. This effect gradually decreased in both groups. In contrast, SN50 alone did not affect TNF-α secretion compared with control cells. Moreover, we observed that TNF-α secretion was significantly higher in the obese hypertensive group than in the overweight group.
The secretion of pro-inflammatory adipokines from adipose tissue has been studied by previous authors [
4,
21,
22]. Adipocytes are known to secrete large quantities of IL-6 as well as non-negligible amounts of TNF-α compared to macrophages [
21,
22], and there is increasing evidence supposing that adipocytes are highly implicated in the inflammatory phenomenon associated with the development of obesity-related hypertension. However, Hoareau et al. [
18] have shown that macrophages are more sensitive to LPS than adipocytes, being able to respond to 5 ng/mL of LPS more than adipocytes, which are sensitive to 50 ng/mL LPS. The number of TLR4 on the surface of the cells could in part explain these differences in response between adipocytes and macrophages [
18]. The production of TNF-α by adipocytes may be of special importance because up to one-third of circulating TNF-α is secreted by adipose tissue [
23]. An increase in central (visceral) adiposity confers a higher metabolic risk. This increased metabolic risk is associated with subclinical inflammation.
This mechanism has been noted in numerous studies, which demonstrate that LPS can stimulate the release of pro-inflammatory cytokines such as TNF-α via NF-κB activation [
6,
18]. They observed that TNF-α production in human adipocytes is dependent on the NF-κB pathway. Lehrke et al. [
6] showed that LPS increases resistin production by inducing secretion of TNF-α. This increase in resistin production can be blocked by both aspirin and rosiglitazone drugs that have a dual anti-inflammatory and insulin-sensitizing action, and they have been shown to antagonize NF-κB activity. Indeed, loss of NF-κB function abolishes LPS induction of resistin [
6]. In a study of adipocytes of mice, it was found that resistin caused insulin resistance and glucose intolerance [
24], and the mice, who were lacking resistin, had low blood glucose levels [
25]. The ability of resistin to modulate glucose metabolism is associated with the activation of SOCS3, an inhibitor of insulin signaling in adipocytes [
24]. Regarding its effect on glucose metabolism, an increase in serum resistin also predicted the risk for increased systolic and diastolic BP in patients with type 2 diabetes mellitus (T2DM) independently of age, gender, BMI, fasting, blood glucose, and HDL-cholesterol [
26].
Zhang et al. [
27] showed that resistin can predict the risk of future hypertension among non-diabetic women aged ≥55 years, even after adjustment for inflammatory and endothelial markers, and can promote endothelial cell activation through the release of ET-1 and up-regulation of VCAM-1 and ICAM-1 [
28]. Resistin was also reported to be decreased with use of probiotics [
29].
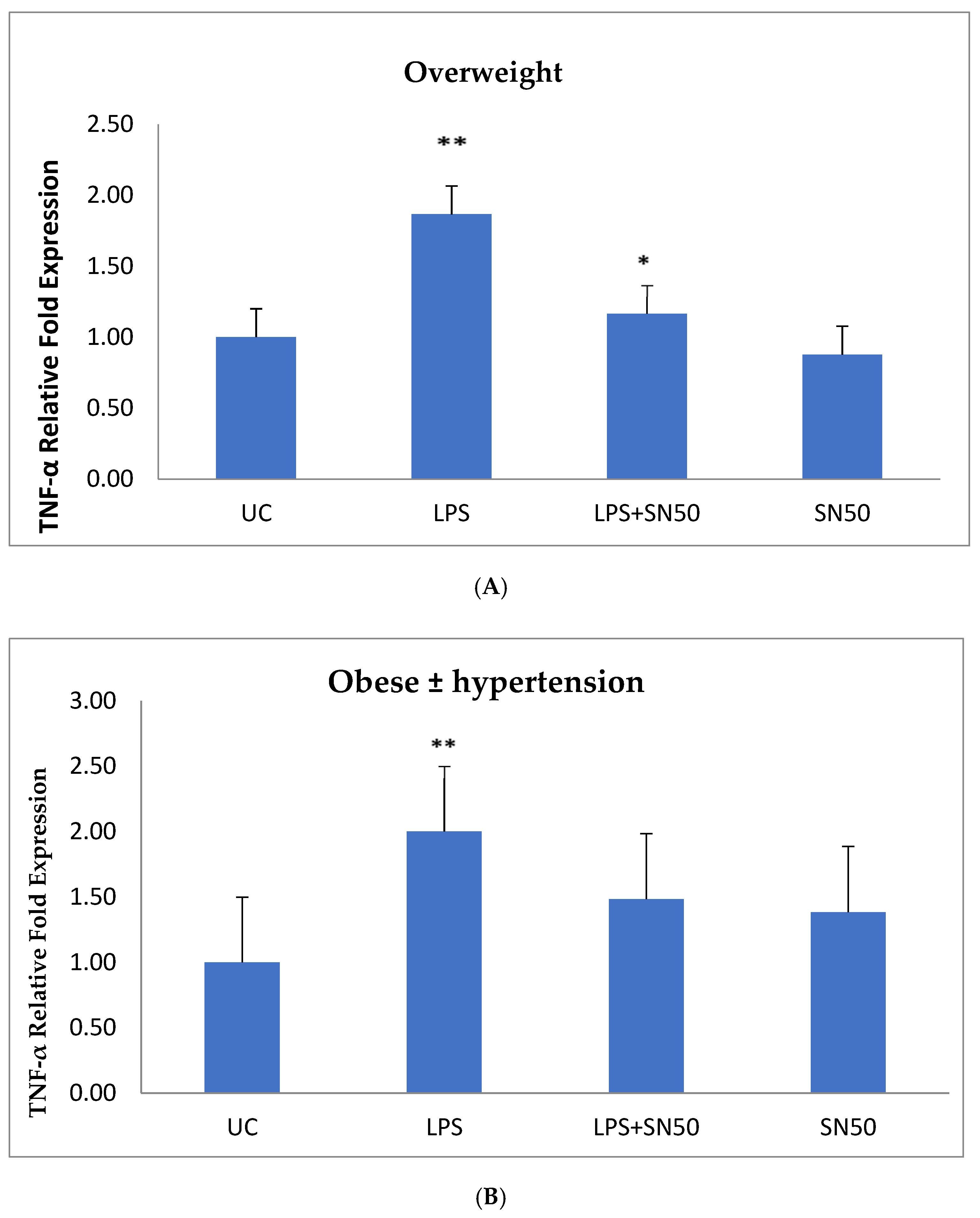
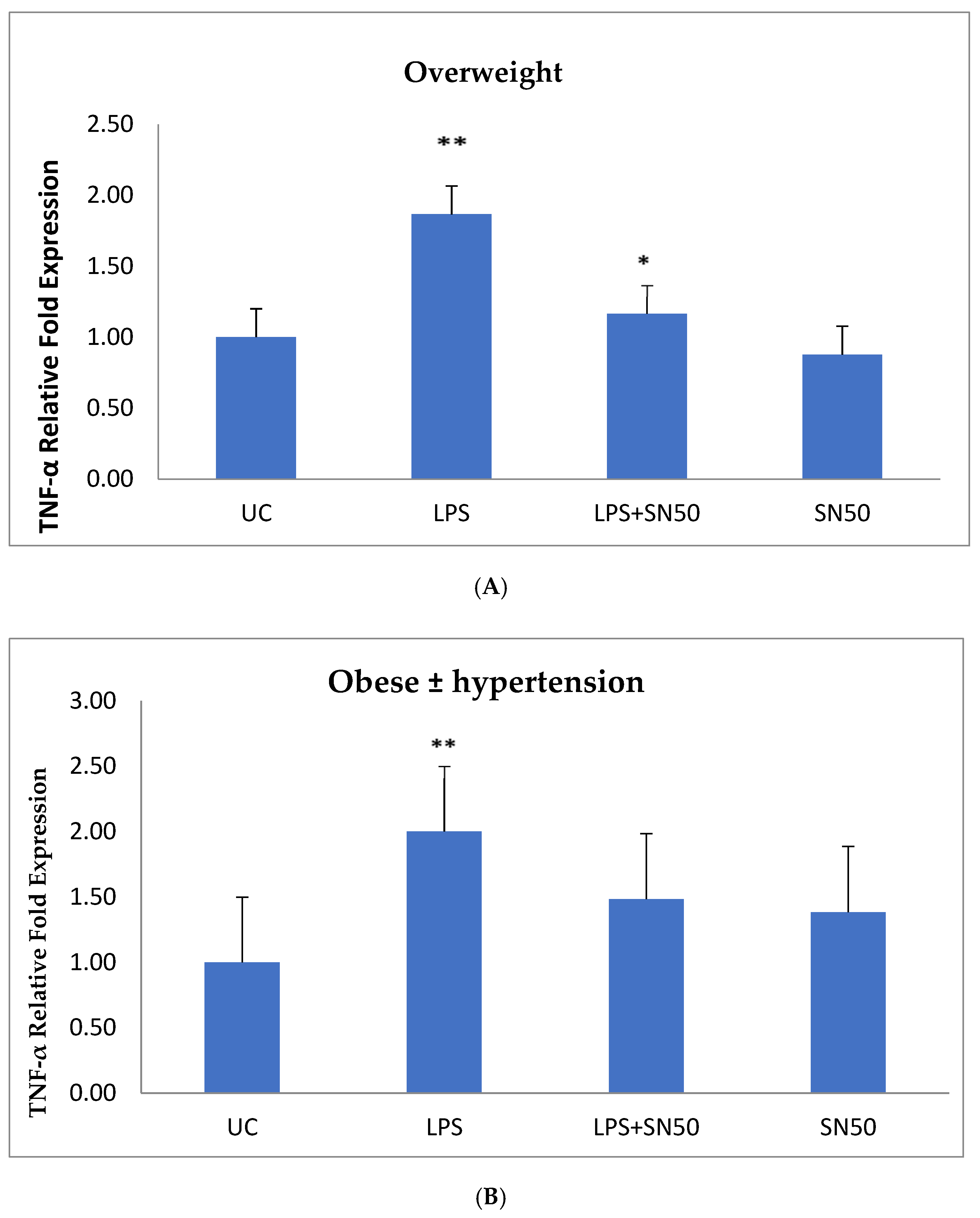
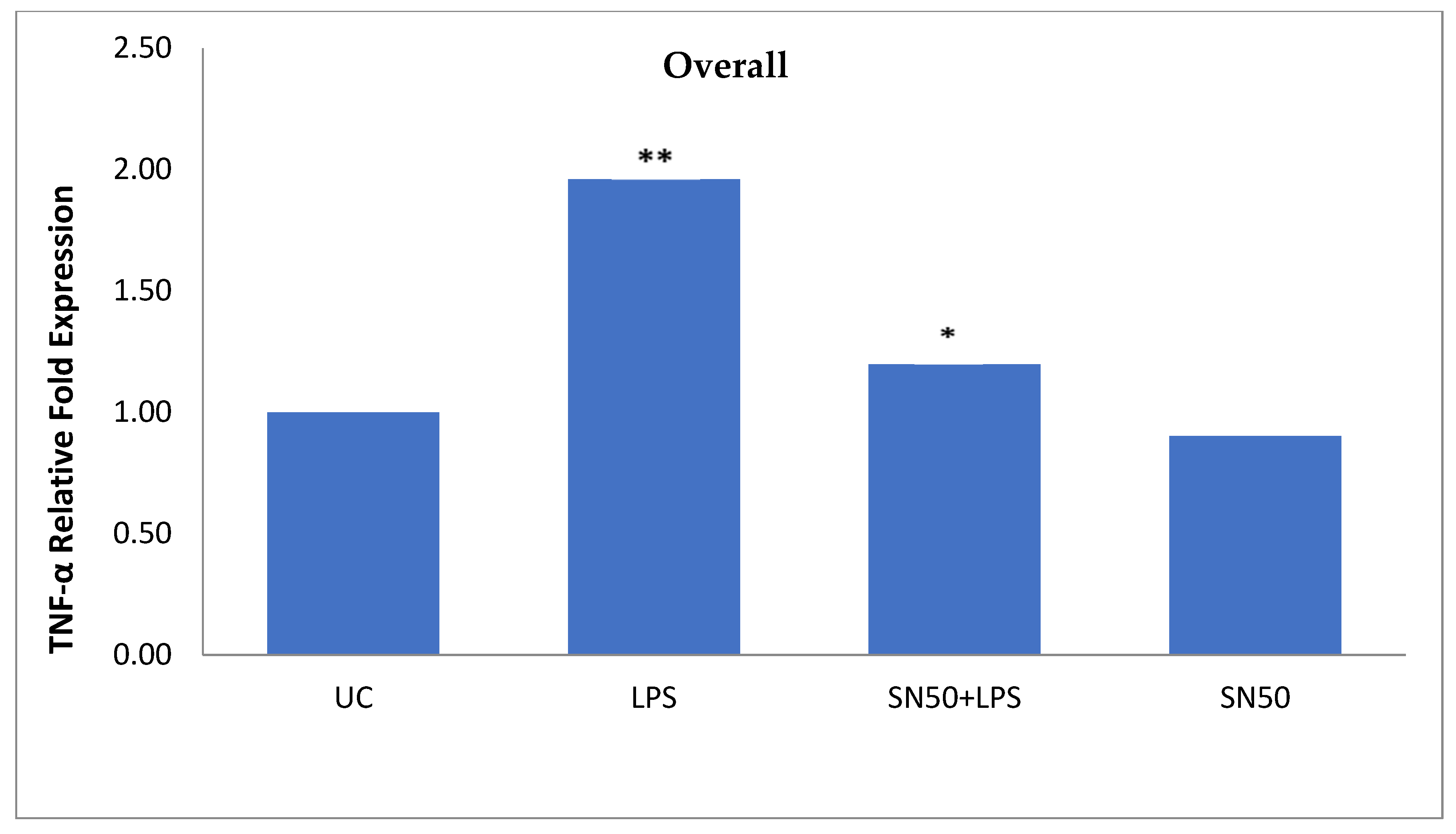
NF-κB plays a role in the regulation of gene transcription, and the present study reported this role in the mRNA TNF-α and AGT levels of overweight patients and obese patients with and without hypertension at 12 h. Treatment of AbdSc adipocytes with LPS increased the mRNA TNF-α level twofold as compared with those in untreated cells in both groups. In contrast, the treatment of AbdSc adipocytes with LPS in the presence of SN50 caused a decrease in the mRNA TNF-α level as compared with those LPS-treated overweight subjects, whereas, no significant change was observed in mRNA TNF-α level in obese subjects with and without hypertension. Moreover, SN50 alone had no effect on TNF-α mRNA levels in both of the observed groups. In contrast, our study demonstrated that the mRNA TNF-α level increased twofold in LPS-treated subjects, as compared with control cells in the combined group, and significantly decreased in the presence of SN50. In addition to its effect on gene expression, LPS also slightly increased the TNF-α protein levels as compared with those in control cells. In contrast, coincubation with LPS and SN50 resulted in a reduction in TNF-α protein levels at 24 h.
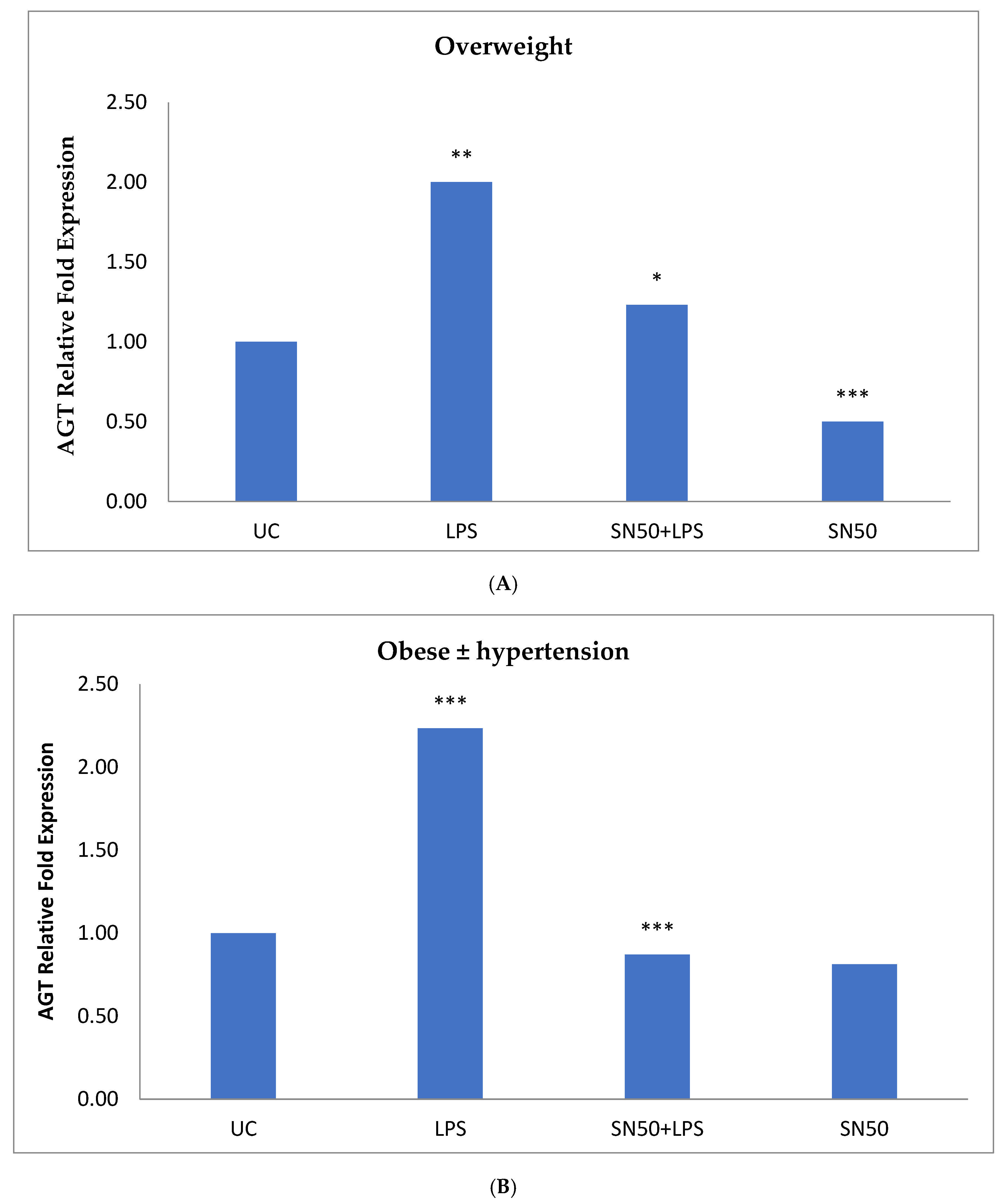
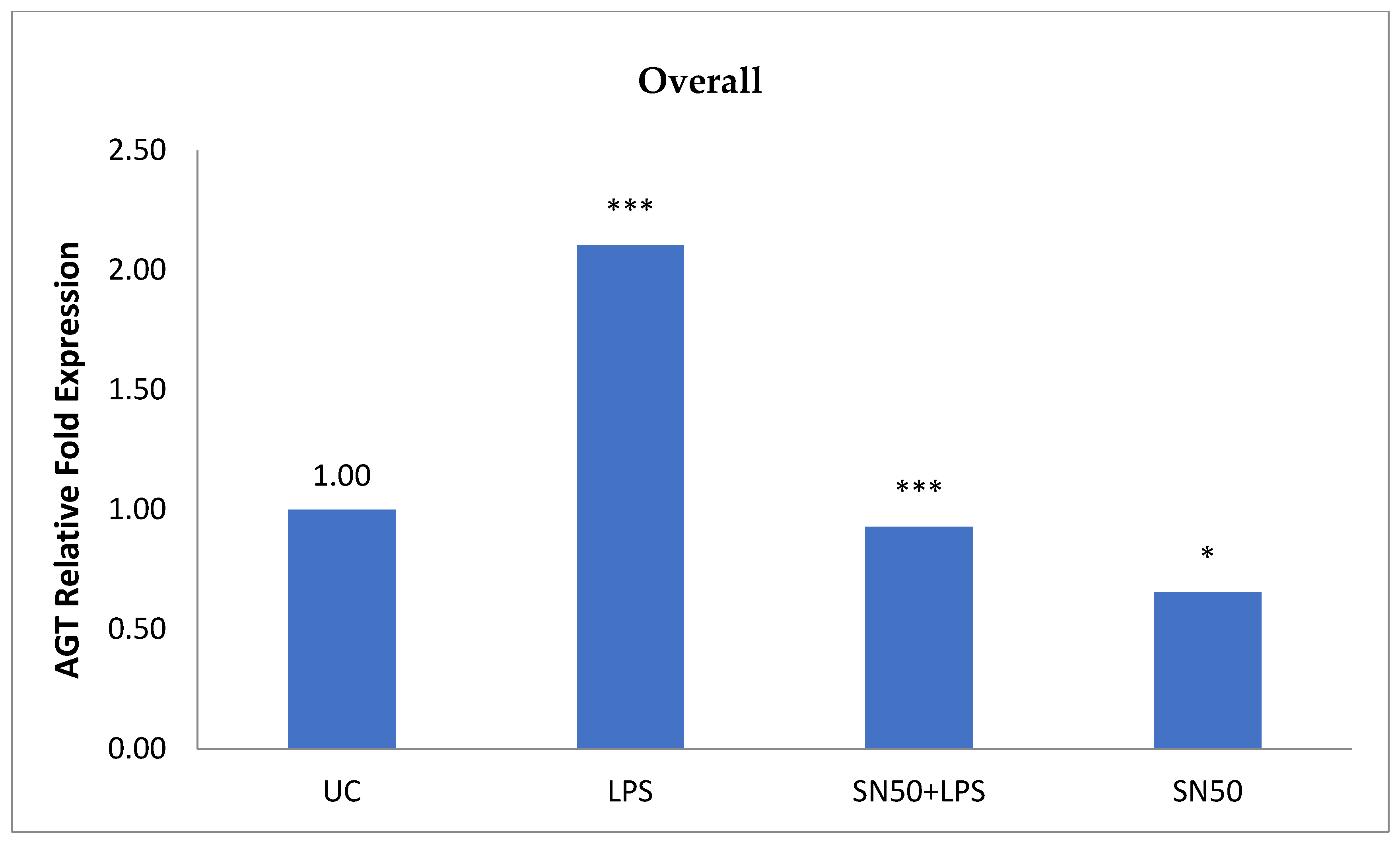
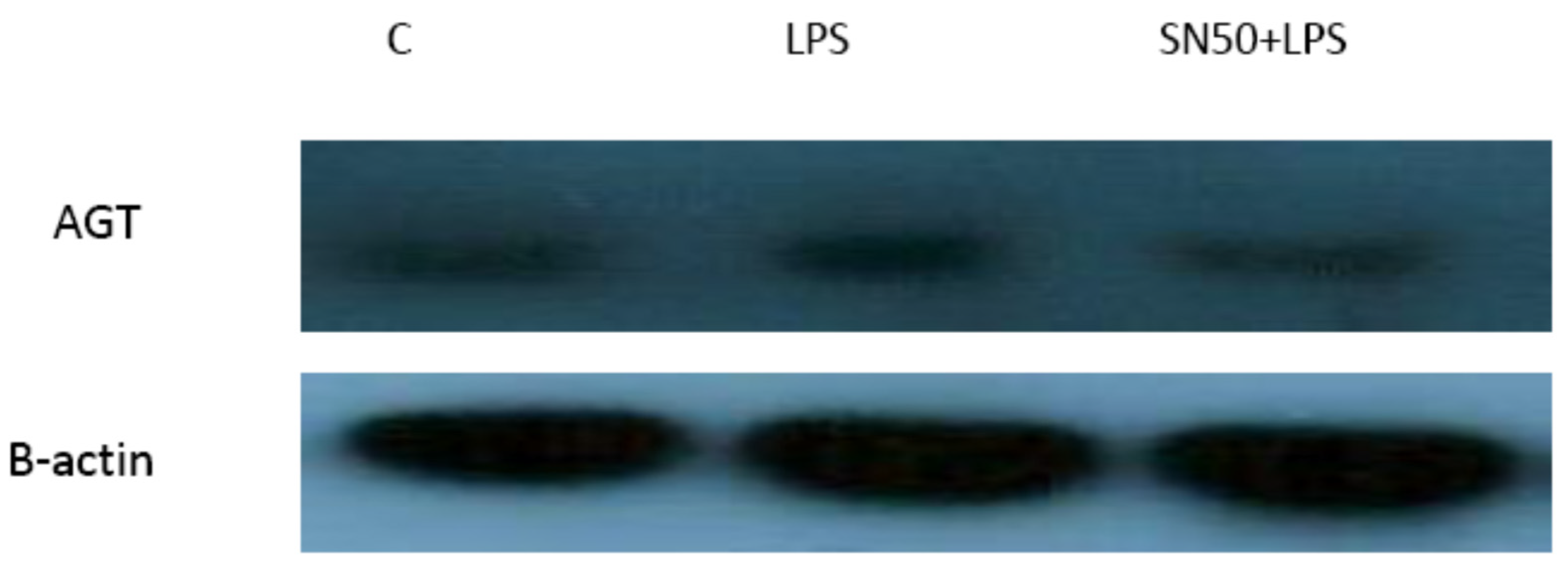
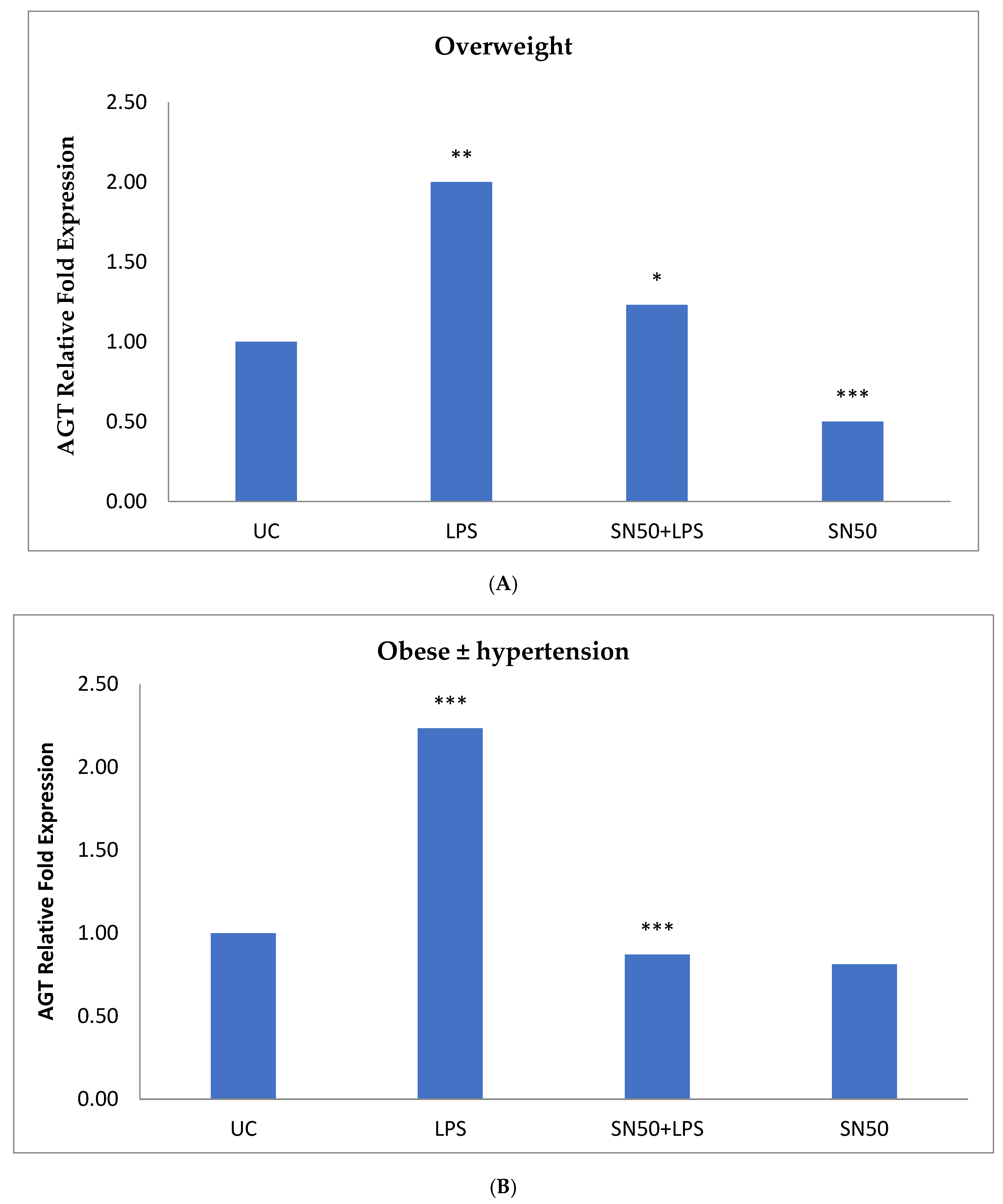
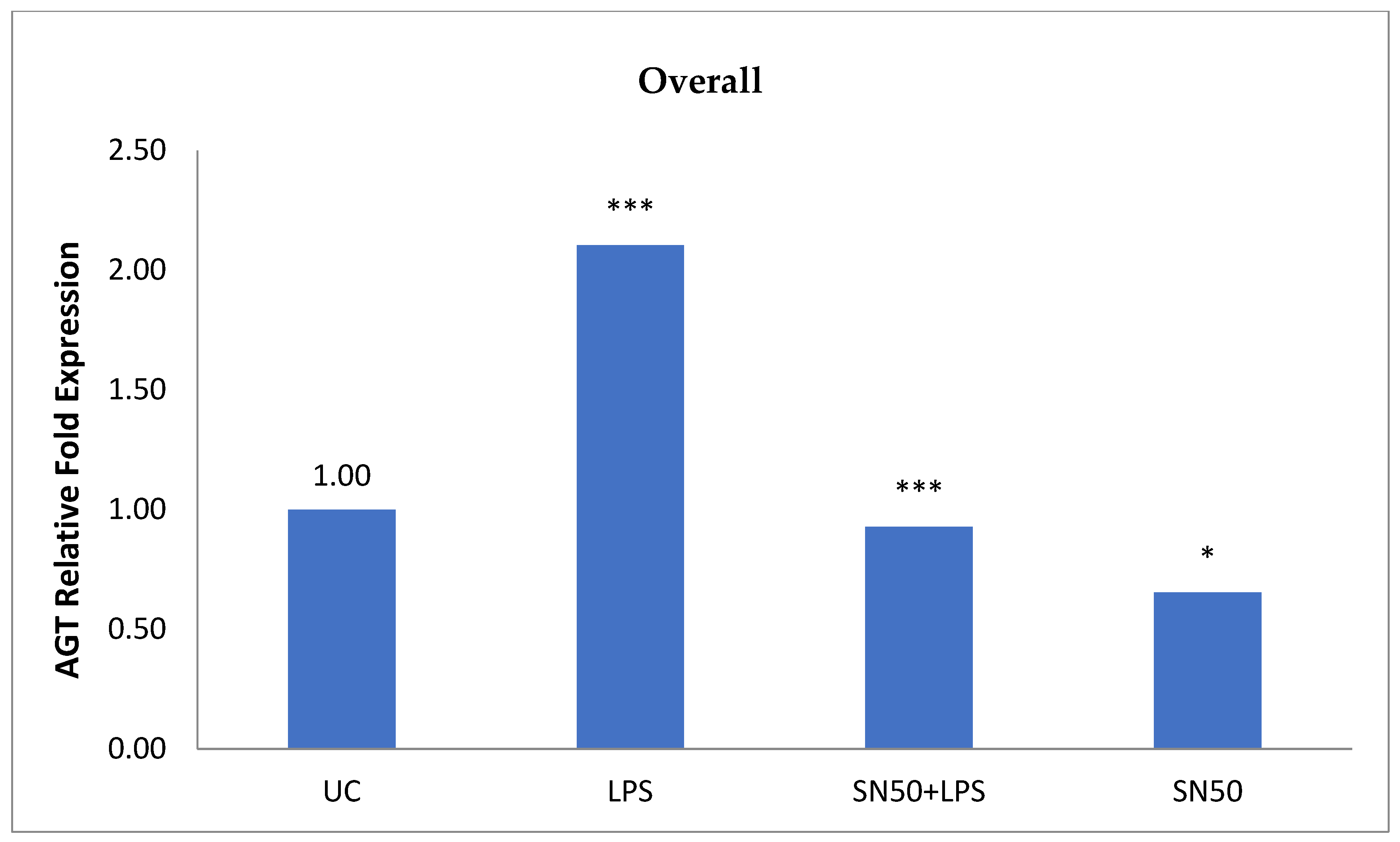
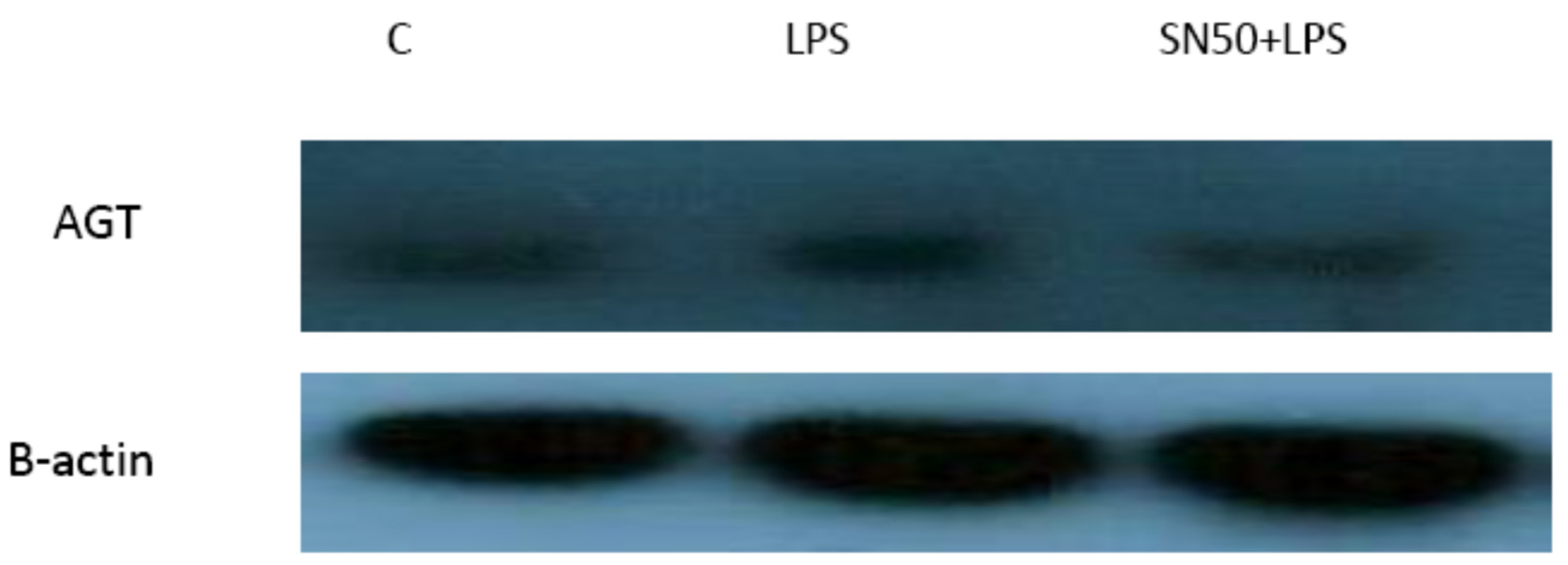
Similarly, treatment of AbdSc adipocytes with LPS caused a twofold increase in the mRNA AGT level as compared with those in control cells in both groups, whereas the treatment of AbdSc adipocytes with LPS in the presence of SN50 caused a decrease in the mRNA AGT level. Moreover, the mRNA AGT was increased twofold in LPS-treated subjects and reduced in the presence of SN50 compared with LPS-treatment in the combined group. LPS alone also slightly increased the AGT protein levels compared with those in control cells. Coincubation with LPS and SN50 resulted in a slight reduction in AGT protein levels at 24 h. Thus, LPS can cause an inflammatory status in adipocytes, and the inflammatory status leads to increased TNF-α via NF-κB and increased AGT and ANG II via RAS. The blocking of NF-κB activation by SN50 led to a decrease in the inflammatory status, which is responsible for obesity and co-morbidities such as hypertension.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}