Thermo/Shear-Responsive Injectable Hydrogels from an Alginate/PNIPAM-Based Graft Copolymer: Effect of Divalent Cations Ca2+ †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experiments
2.1. Materials
2.2. Synthesis of the Graft Copolymer NaALG-g-P(NIPAM94-co-NtBAM6)-NH2
2.3. Hydrogels Preparation
2.4. Rheological Studies
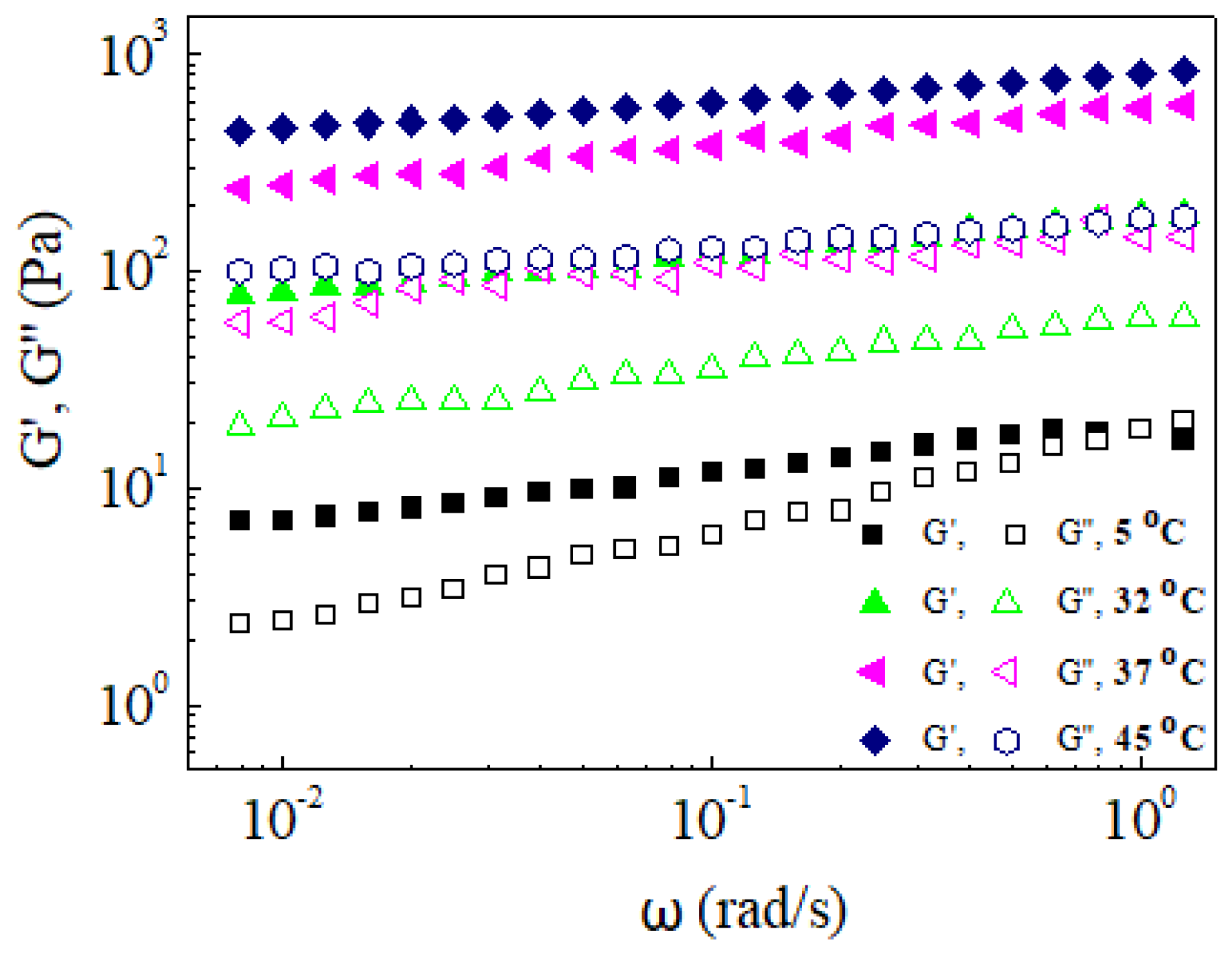
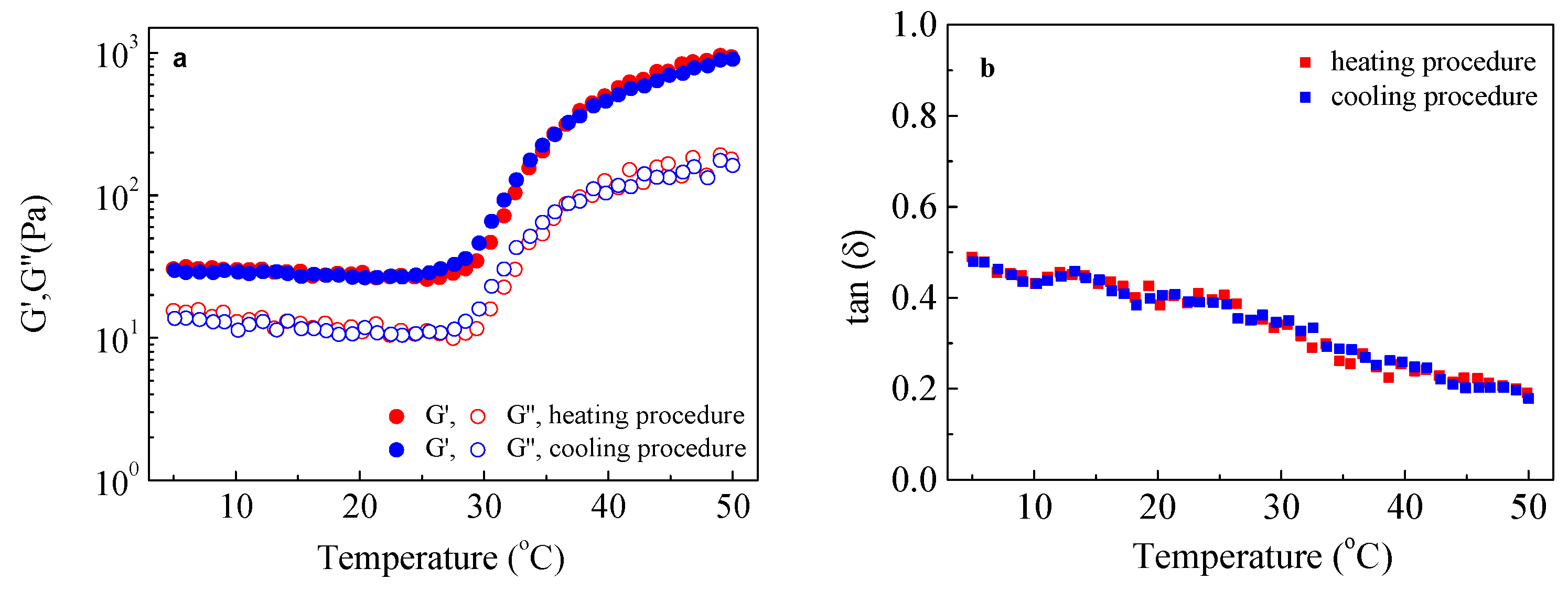
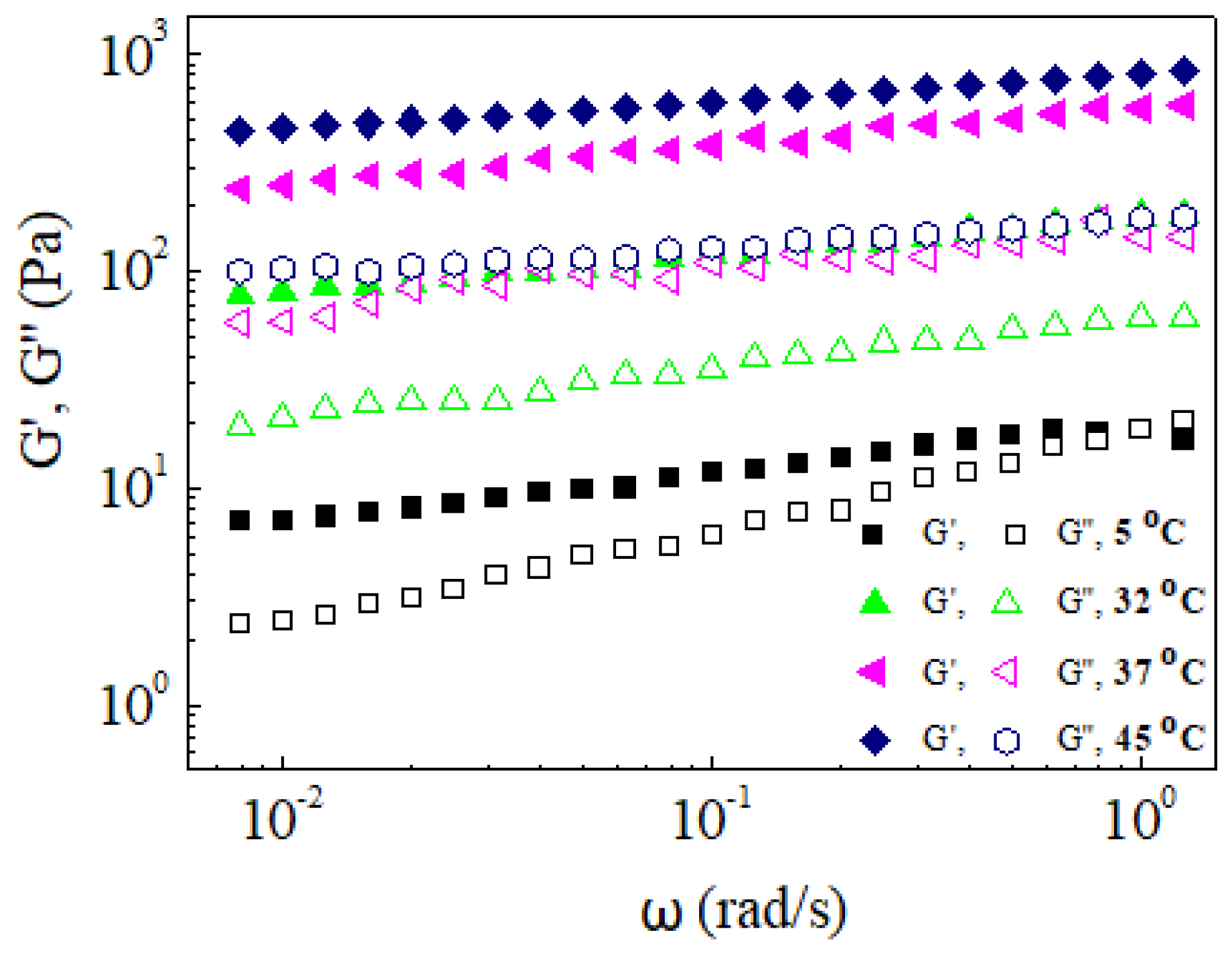
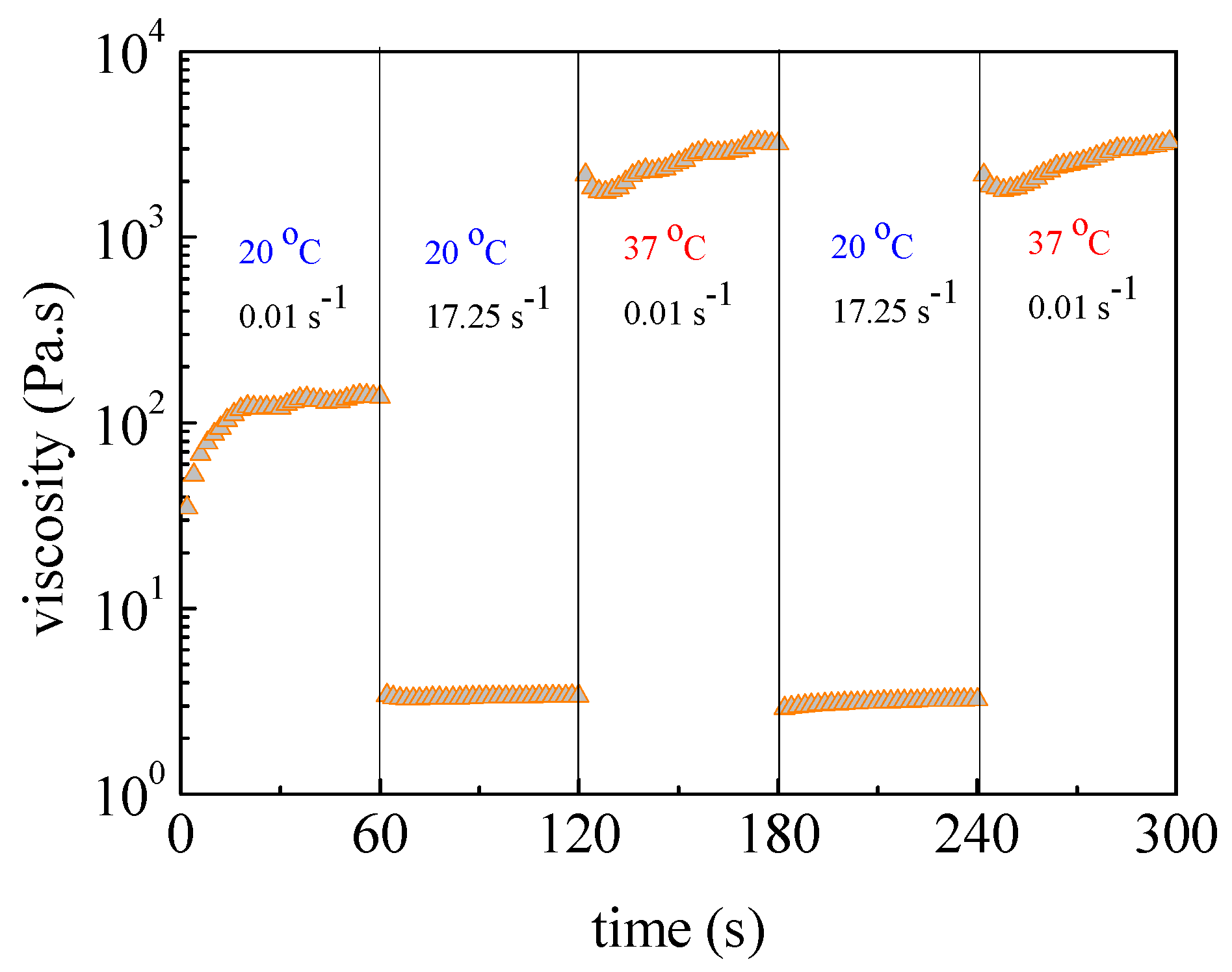
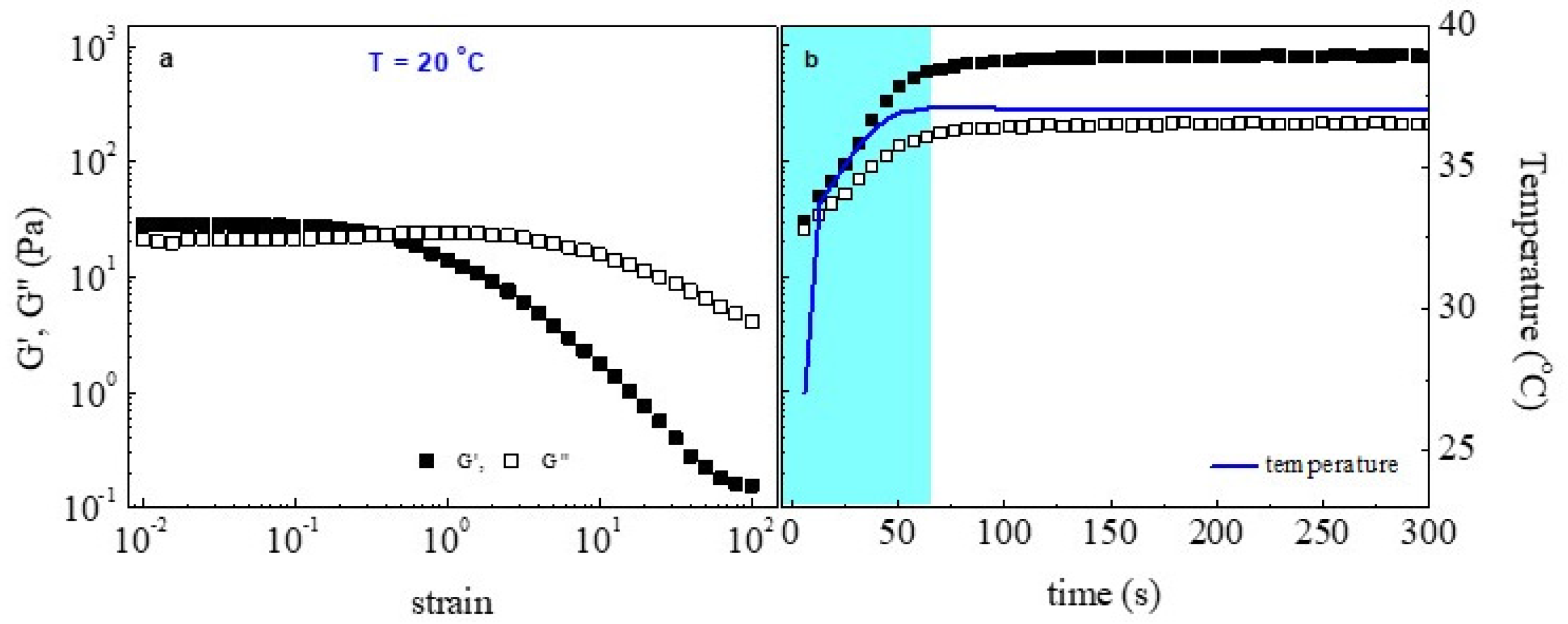
3. Results
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| NaALG | Sodium Alginate |
| PNIPAM | Poly(N-isopropylacrylamide) |
| NtBAM | N-tertiary-butyl-acrylamide |
| NIPAM | N-isopropylacrylamide |
| KPS | Potassium Peroxodisulfate |
| AET HCl | 2-Aminoethanethiol Hydrochloride |
| EDC | 1-Ethyl-3-(3-(dimethylamino) propyl) Carbodiimide |
| HOBt | 1-Hydroxybenzotriazole Hydrate |
| DMF | Dimethylformamide |
| HCl | Hydrochloric Acid |
| NaOH | Sodium Hydroxide |
| CaCl2 H2O | Calcium Chloride dihydrate |
References
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Leal, D.; de Borggraeve, W.; Encinas, M.V.; Matsuhiroa, B.; Müller, R. Preparation and characterization of hydrogels based on homopolymeric fractions of sodium alginate and PNIPAAm Responsive reversible hydrogels from associative ‘‘smart’’ macromolecules. Carbohydr. Polym. 2013, 92, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.G.; Rinaudo, M.; Villar, M.A. Oxidation of sodium alginate and characterization of the oxidized derivatives. Carbohydr. Polym. 2007, 67, 296–304. [Google Scholar] [CrossRef]
- Yang, Y.; Campanella, O.H.; Hamaker, B.R.; Zhang, G.; Gu, Z. Rheological investigation of alginate chain interactions induced by concentrating calcium cations. Food Hydrocoll. 2013, 30, 26–32. [Google Scholar] [CrossRef]
- Grant, G.T.; Morris, E.R.; Rees, D.A.; Smith, P.J.; Thom, D. Biological interactions between polysaccharides and divalent cations: The egg-box model. FEBS Lett. 1973, 32, 195–198. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, G.; Tsitsilianis, C. LCST Polymers: Thermoresponsive Nanostructured Assemblies towards Bioapplications. Polymer 2020, 211, 123146. [Google Scholar] [CrossRef]
- Iatridi, Z.; Saravanou, S.F.; Tsitsilianis, C. Injectable self-assembling hydrogel from alginate grafted by P(Nisopropylacrylamide-co-N-tert-butylacrylamide) random copolymers. Carbohydr. Polym. 2019, 219, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Ciocoiua, O.N.; Staikosa, G.; Vasile, C. Thermoresponsive behavior of sodium alginate grafted with poly(Nisopropylacrylamide) in aqueous media. Carbohydr. Polym. 2018, 184, 118–126. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saravanou, S.-F.; Kounelaki, F.; Tsitsilianis, C. Thermo/Shear-Responsive Injectable Hydrogels from an Alginate/PNIPAM-Based Graft Copolymer: Effect of Divalent Cations Ca2+. Proceedings 2021, 69, 28. https://doi.org/10.3390/CGPM2020-07196
Saravanou S-F, Kounelaki F, Tsitsilianis C. Thermo/Shear-Responsive Injectable Hydrogels from an Alginate/PNIPAM-Based Graft Copolymer: Effect of Divalent Cations Ca2+. Proceedings. 2021; 69(1):28. https://doi.org/10.3390/CGPM2020-07196
Chicago/Turabian StyleSaravanou, Sofia-Falia, Fotoula Kounelaki, and Constantinos Tsitsilianis. 2021. "Thermo/Shear-Responsive Injectable Hydrogels from an Alginate/PNIPAM-Based Graft Copolymer: Effect of Divalent Cations Ca2+" Proceedings 69, no. 1: 28. https://doi.org/10.3390/CGPM2020-07196
APA StyleSaravanou, S.-F., Kounelaki, F., & Tsitsilianis, C. (2021). Thermo/Shear-Responsive Injectable Hydrogels from an Alginate/PNIPAM-Based Graft Copolymer: Effect of Divalent Cations Ca2+. Proceedings, 69(1), 28. https://doi.org/10.3390/CGPM2020-07196