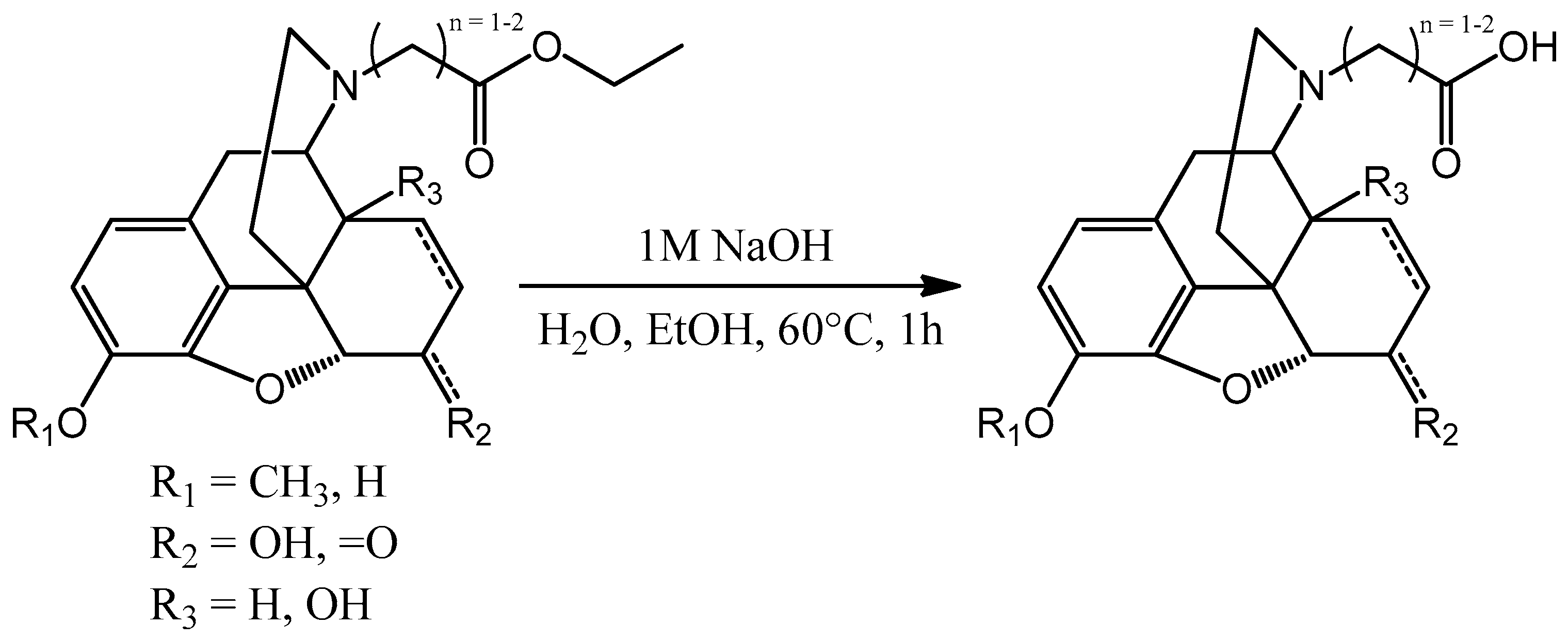1. Introduction
Drug abuse is a worldwide problem. Even today one of the most popular illegal substance is morphine and its derivative heroin beside cocaine and marihuana. These drugs have a very serious potent to cause damage not just to the individual but to the whole society as well [
1,
2].
To help the drug addicts—and now let’s just focus on the opiate users—there isn’t a lot of possibilities. The most common way is the methadone therapy. In this case the morphine or heroin is substituted with methadone because its half-life is longer then the illegal drugs. Therefore, the dosage of the morphine and heroin can be decreased until it reaches zero. After this it is possible to decrease the level of methadone as well without inducing the withdrawal effects of the opiates. Theoretically this protocol works fine but in practice it can be easily spoiled. Namely if the patient uses the illegal substance during the therapy all of the results that are achieved so far are just a waste of time, money and energy. Not even mention that the level of tolerance decreases as well and that can cause the death of the patient too.
For this problem an alternative solution can be the vaccination against these drugs [
3]. Drugs of abuse are small molecules that typically do not induce an antibody response following the administration. To induce antibodies against these kind of molecules, structural changes have to be made to obtain so called “haptens”. The hapten must be coupled to immunogenic proteins, called “carriers”. These connected derivatives are typically drug-linker adducts, in which the linker has a terminal functional group (i.e., carboxylic acid or aliphatic amine) that forms a covalent bond with the carrier. The efficacy of these conjugate vaccines depends on several factors including hapten design, coupling strategy, hapten density, carrier protein selection, and vaccine adjuvant [
4,
5].
2. Materials and Methods
To obtain a hapten molecule the morphine derivatives can be modified on different sites:
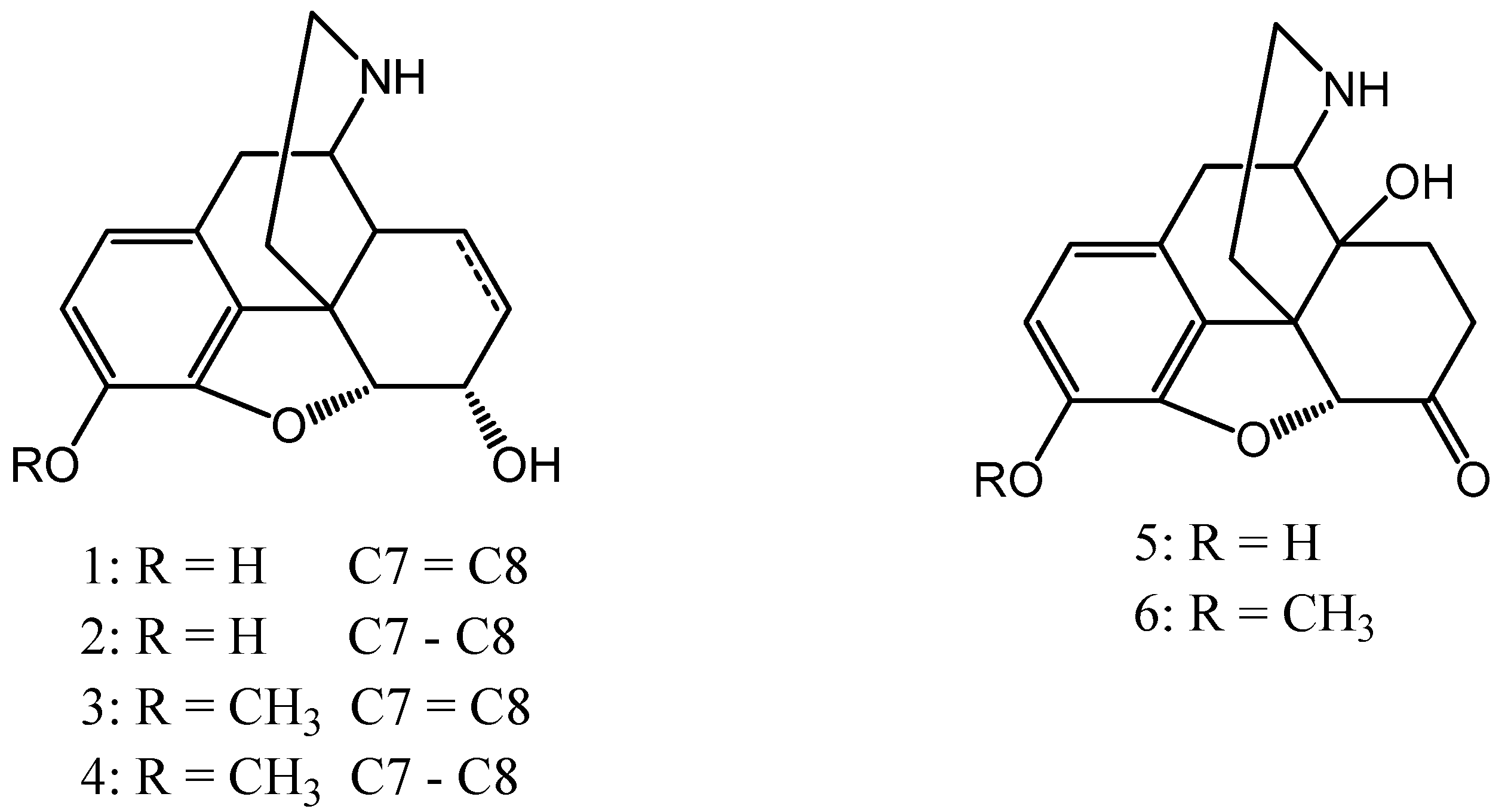
For our purpose the desired normorphine derivatives (normorphine (1), dihydronormorphine (2), norcodeine (3), dihydronorcodein (4), noroxymorphone (5) and noroxycodone (6) (
Figure 1)) could be obtained by the
N-demethylation of the starting molecules [
6,
7,
8,
9,
10,
11,
12].
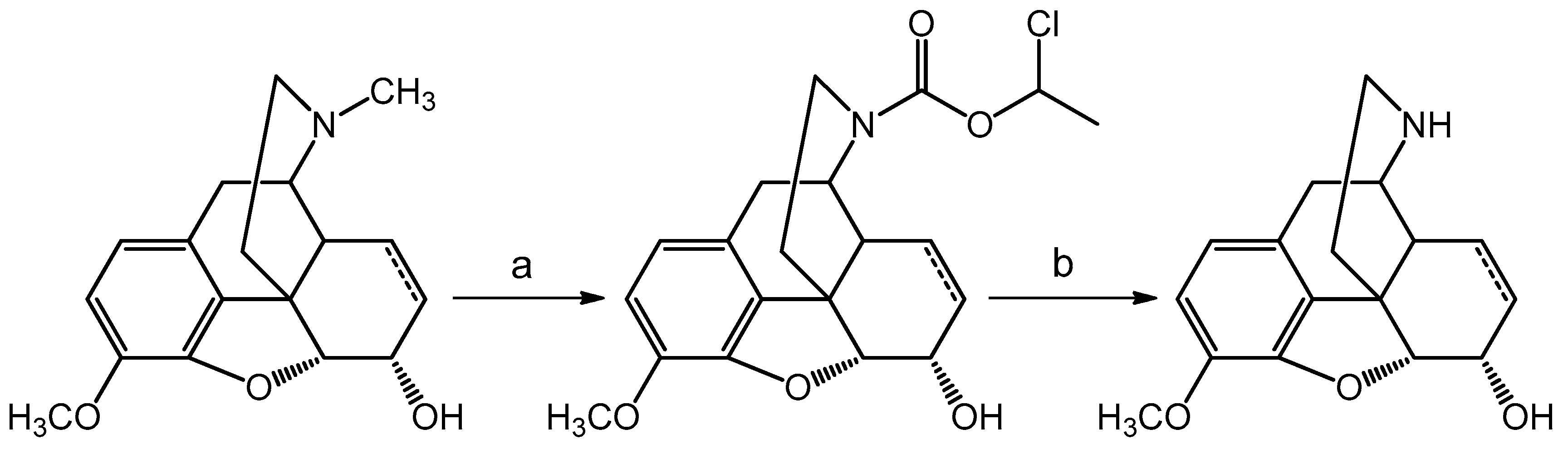
Codeine or dihydrocodeine can be
N-demethylated with α-chloro-ethyl chloroformate in 1,2-dichloroethane solvent and the intermediate carbamate was heated with methanol to yield the hydrochloride salt of norcodeine (dihydronorcodeine) (
Scheme 1).
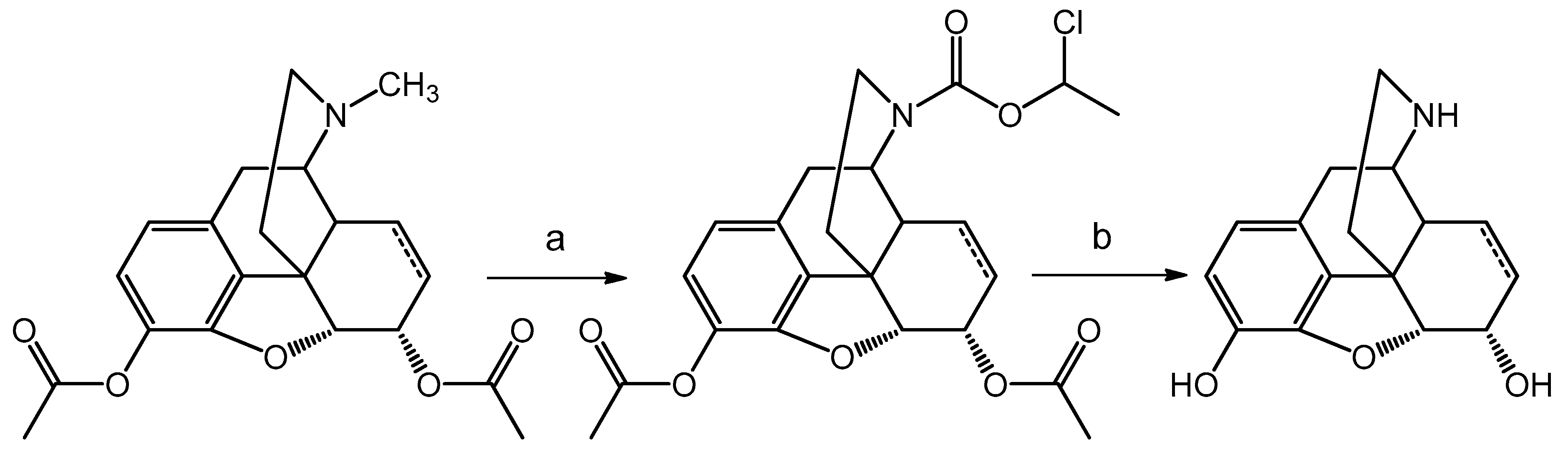
For the preparation of (dihydro)normorphine 3,6-diacetyl (dihydro)morphine was
N-demethylated with α-chloro-ethyl chloroformate and after cleavage of the carbamate 3,6-diacetyl (dihydro)normorphine hydrochloride salt was obtained. Acid hydrolysis of 3,6-diacetyl (dihydro)normorphine yielded (dihydro)normorphine (
Scheme 2).
3,14-di-
O-acetyloxymorphone and 14-
O-acetyloxycodone were
N-demethylated by the above procedure, and these reactions resulted in 3,14-di-
O-acetyloxynormorphone hydrochloride and 14-
O-acetylnoroxycodone hydrochloride. Acid hydrolysis (10% HCl, 6 h reflux) yielded noroxymorphone and noroxycodone (
Scheme 3).
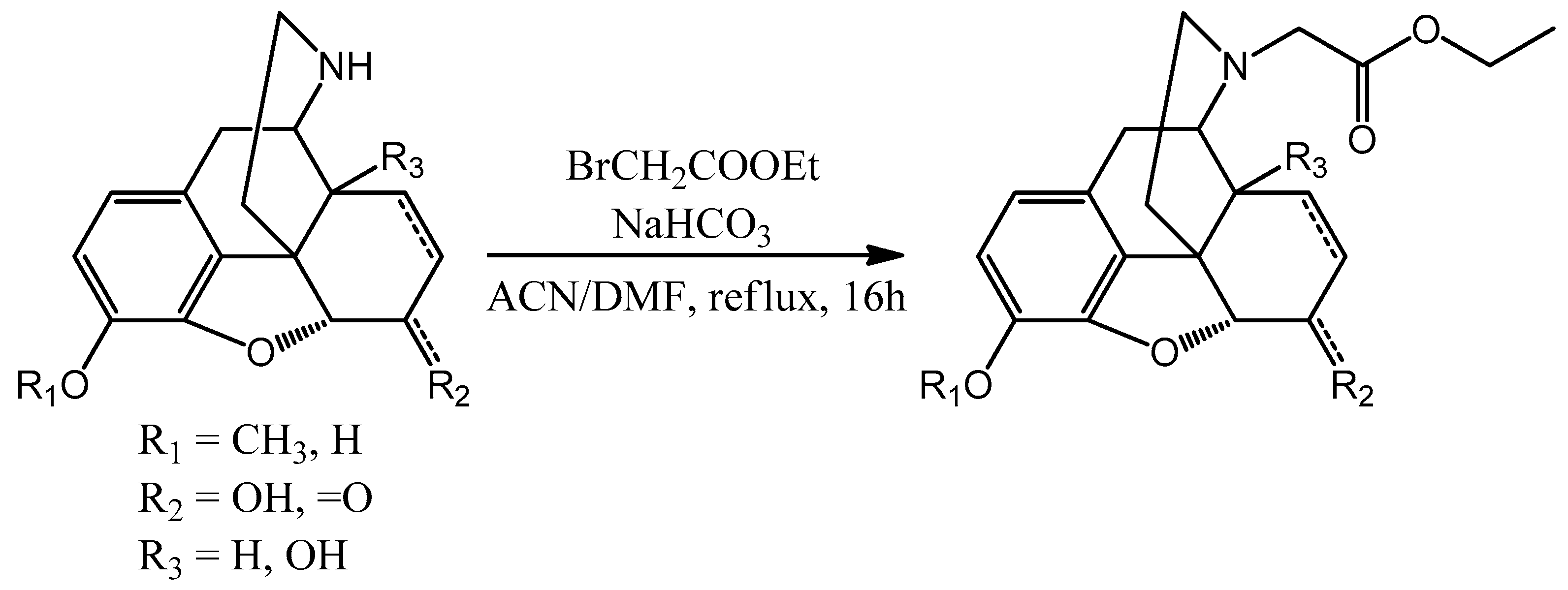
The next step is the N-alkylation of the norcompounds. Depending on the linker chain size different methods were used.
In the case of methylene bridge ethyl bromoacetate was used (
Scheme 4). Depending on the solubility the starting nor-compound was dissolved in dimethyl formamide (DMF) or acetonitrile (ACN). In the presence of sodium bicarbonate, the reaction mixture was refluxed for 16 h. The conversion of the reaction was monitored by thin layer chromatography (TLC). The inorganic salts were filtered and in the case of acetonitrile it was evaporated in vacuo. The DMF was vacuum distillated and water was added to the residue. The aqueous phase was basified with cc. ammonia and was extracted with chloroform. The organic phase was dried over sodium sulfate. The solution was filtered and the chloroform was evaporated to obtain the specific
N-carboxymethyl-nor-compound ethyl ester.
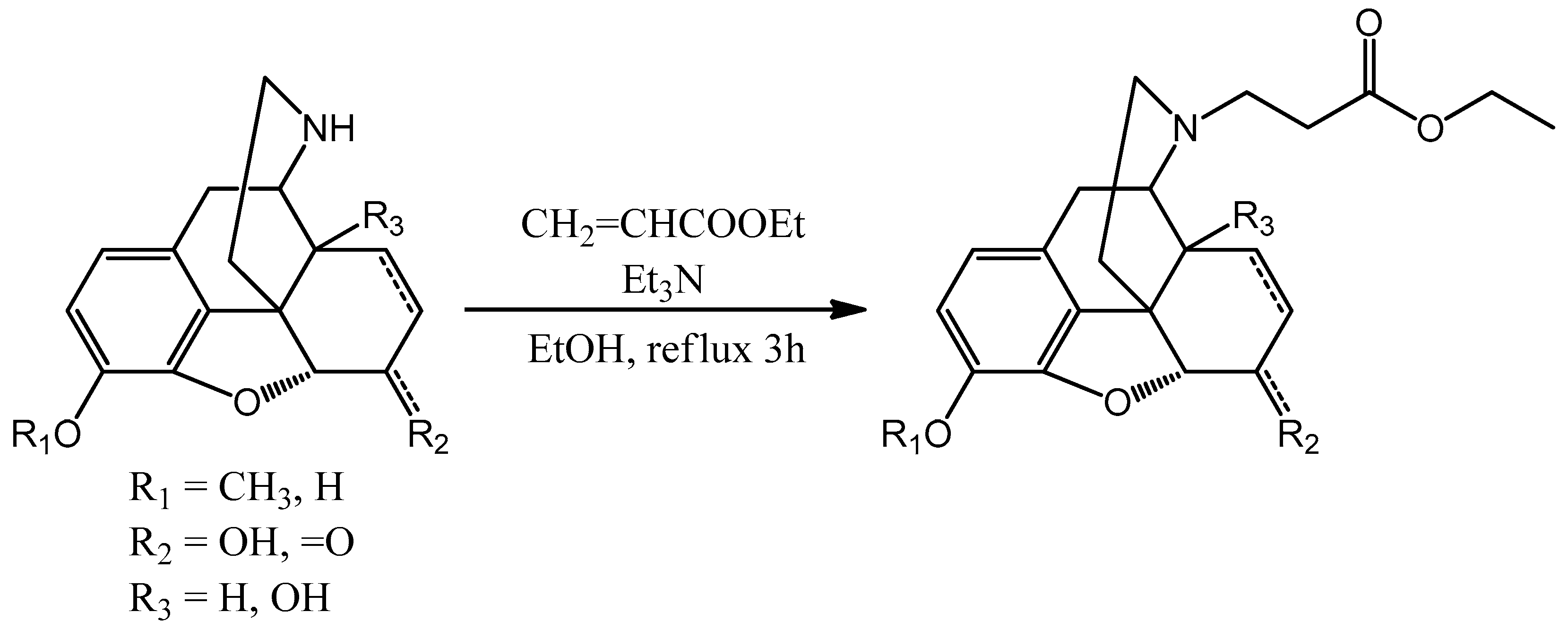
For the ethylene linker ethyl acrylate was utilized (
Scheme 5). The nor-molecule was dissolved in ethanol and in the presence of triethylamine the mixture was refluxed for 3 h. After checking the TLC, the reaction mixture was evaporated in vacuo. The product was in every case the
N-carboxyethyl-nor-compound ethyl ester.
After obtaining of the aforementioned esters all of them were hydrolyzed (
Scheme 6). Sodium hydroxide was used in every case, the reaction mixture was heated up to 60 °C and was stirred for 30–60 min. TLC was used to monitor the reaction. After the conversion the pH was set to 3–4 and the mixture was evaporated to dryness to get the hydrochloride salt.
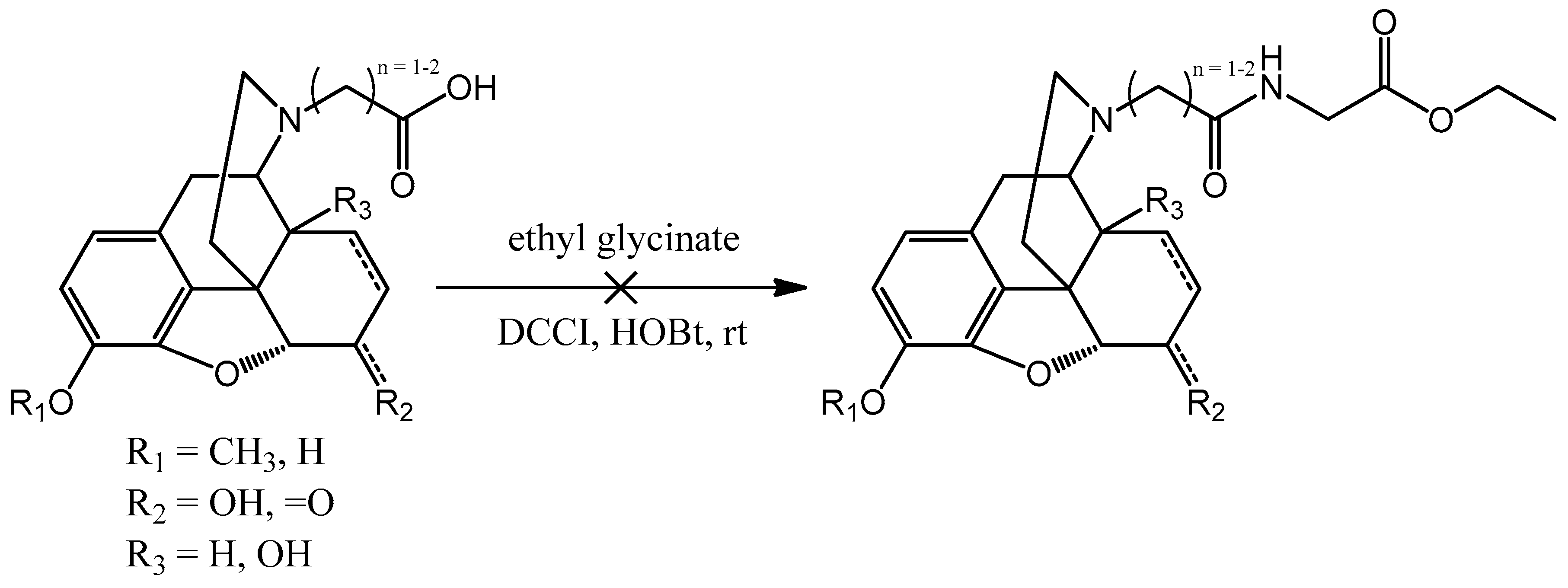
In the next step the carboxylic group containing molecules were supposed to react with glycine ethyl ester (
Scheme 7). Reagents that are common in peptide syntheses were used like
N,
N′-dicyclohexyl carbodiimide (DCCI) or 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDAC) and 1-hydroxybenzotriazole (HOBt). Unfortunately, these connections were very low yielding reactions and the mixtures did not result the desired compounds. To obtain the glycine derivatives an alternative route has been chosen.
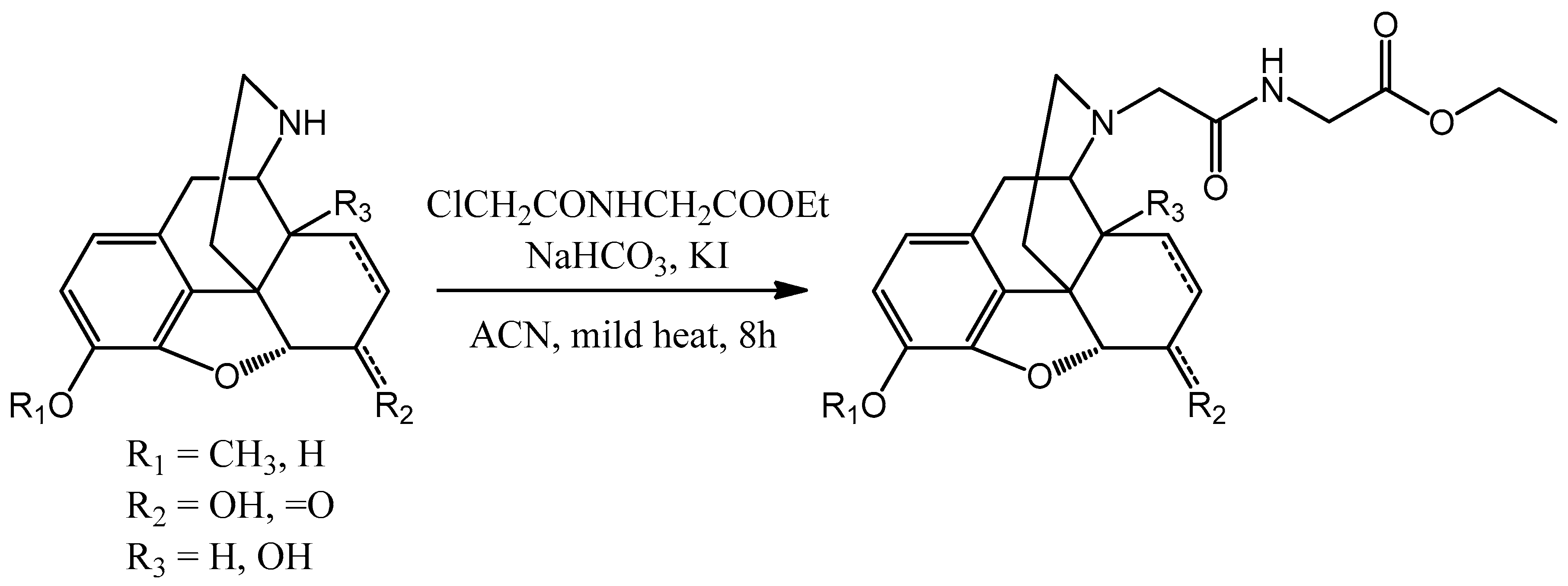
In the case of methylene bridge containing compounds the retro-synthetic analysis revealed that the linker between the nor-compounds and the glycine ethyl ester could be the chloroacetyl chloride. Because of this
N-chloroacetly-glycine ethyl ester was a good choice as the reagent (
Scheme 8).
The nor-compound was dissolved in acetonitrile. In the presence of potassium iodide and sodium bicarbonate it was reacted with N-chloroacetly-glycine ethyl ester. The mixture was heated and stirred until the conversion was complete. After the evaporation water and concentrated ammonia was added to the residue and it was extracted with chloroform. The organic phase was dried on sodium sulfate and after the evaporation of the organic solvent the desired product was obtained. Column chromatography was used if purification was needed.
The esters from the previous reaction were hydrolyzed as mentioned above after reacting with ethyl bromoacetate and ethyl acrylate.
3. Results
Totally 36 compounds have been synthetized and 32 of them are brand new molecules (
N-carboxymethyl-normorphine ethyl ester,
N-carboxyethyl-normorphine ethyl ester,
N-carboxyethyl-normorphine,
N-carboxyethyl-norcodein were previously synthesized) [
13,
14]. All of them are under physico-chemical examination and 8 of them (
N-acetylglycine-normorphine ethyl ester,
N-acetylglycine-dihydronormorphine ethyl ester,
N-acetylglycine-noroxymorphone ethyl ester,
N-acetylglycine-noroxycodone ethyl ester,
N-acetylglycine-normorphine,
N-acetylglycine-dihydronormorphine,
N-acetylglycine-noroxymorphone,
N-acetylglycine-noroxycodone) are sent for biological and animal experiments.
3.1. N-demethylation Reactions
Codeine or dihydrocodeine (3 mmol) was dissolved in dried 1,2-dichloroethane (30 mL) then sodium hydrogen carbonate (0.84 g) was added to the solution. To this ice-cold mixture α-chloroethyl chloroformate (10 mmol) was added dropwise and the reaction mixture was stirred at room temperature for 30 min. then heated at 90 °C overnight. The resulting suspension was cooled to room temperature and inorganic salts were filtered off. The filtrate was evaporated under reduced pressure and the residue was dissolved in methanol and the solution was heated at 60 °C for 6 h. Methanol was removed under reduced pressure to yield a crystalline solid, the hydrochloride salt of norcodeine or dihydronorcodeine. Free base was liberated with 10% sodium hydroxide (pH = 9) and was extracted with chloroform. The chloroform extract was dried (sodium sulfate) and evaporated to result in the norcompound.
(Dihydro)normorphine was prepared by N-demethylation of diacetyl (dihydro)morphine by means of α-chloroethyl chloroformate using the above-mentioned procedure and the carbamate was treated with methanol at 60 °C to afford diacetyl (dihydro)normorphine hydrochloride salt. (Dihydro)normorphine was obtained by hydrolysis of diacetyl (dihydro)normorphine x HCl in 5% HCl at 100 °C for 4 h. (Dihydro)normorphine base was precipitated with 25% ammonia solution (pH = 9–10) and filtered off.
14-O-Acetyloxycodone (2 mmol) was N-demethylated with α-chloroethyl chloroformate (10 mmol) for 16 h. The reaction was monitored by thin-layer chromatography to check the conversion, if it was necessary another 5 mmol portion of α-chloroethyl chloroformate was added. The carbamate intermediate was decomposed in methanol to yield the hydrochloride salt of 14-O-acetylnoroxycodone. Acid hydrolysis in refluxing 10% HCl for 6 h resulted in the hydrochloride salt of noroxycodone. The free base of noroxycodone was liberated from the acid solution 10% sodium hydroxide (pH = 10) and it was extracted with chloroform. The chloroform extract was dried under sodium sulfate and after evaporation of the solution afforded the oily noroxycodone, which was rubbed with diethyl ether to produce crystalline material.
N-demethylation of 3,14-di-O-acetyloxymorphone yielded the hydrochloride salt of 3,14-di-O-acetylnoroxymorphone, which was hydrolyzed in 10% HCl solution. Noroxymorphone base was precipitated from the acid solution with 25% ammonia solution to yield noroxymorphone which is pure for further reactions.
3.2. General Synthesis of N-carboxymethyl-nor-compound Ethyl Esters
The specific normorphine derivative (2 mmol) was dissolved in 30 mL of acetonitrile. In the presence of sodium bicarbonate (10 mmol) ethyl bromoacetate (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 16 h. After the TLC showed no more starting compound the inorganic salts were filtered and the solvent was evaporated. Water (30 mL) was added to the residue and the pH was set around 9 with conc. ammonia. Then it was extracted with chloroform (3 × 25 mL) and after unifying the organic phases it was dried on sodium sulfate. After filtration the chloroform was evaporated. If it was necessary column chromatography was used. (Choloroform: Methanol 9:1)
N-carboxymethyl-normorphine ethyl ester yield: 86%
N-carboxymethyl-dihydronormorphine ethyl ester yield: 84%
N-carboxymethyl-norcodeine ethyl ester yield: 90%
N-carboxymethyl-dihydronorcodeine ethyl ester yield: 88%
N-carboxymethyl-noroxymorphone ethyl ester yield: 82%
N-carboxymethyl-noroxycodone ethyl ester yield: 87%
3.3. General Synthesis of N-carboxymethyl-nor-compounds
The specific N-carboxymethyl-nor-compound ethyl ester (0.5 mmol) was added to the mixture of ethanol (1 g) and water (2.5 mL). The mixture was reacted with sodium hydroxide (1 M, 0.5 mmol) for 1 h at 60 °C. After the reaction the pH was set to 3–4 with 10% HCl solution. Then the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
N-carboxymethyl-normorphine yield: 92%
N-carboxymethyl-dihydronormorphine yield: 93%
N-carboxymethyl-norcodeine yield: 95%
N-carboxymethyl-dihydronorcodeine yield: 90%
N-carboxymethyl-noroxymorphone yield: 96%
N-carboxymethyl-noroxycodone yield: 94%
3.4. General Synthesis of N-carboxyethyl-nor-compound Ethyl Esters
The specific normorphine derivative (2 mmol) was dissolved in 30 mL of ethanol. In the presence of triethylamine (1 mL) ethyl acrylate (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 3 h. After the TLC showed no more starting compound the solvent was evaporated. If it was necessary column chromatography was used. (Choloroform: Metahnol 9:1)
N-carboxyethyl-normorphine ethyl ester yield: 98%
N-carboxyethyl-dihydronormorphine ethyl ester yield: 96%
N-carboxyethyl-norcodeine ethyl ester yield: 97%
N-carboxyethyl-dihydronorcodeine ethyl ester yield: 93%
N-carboxyethyl-noroxymorphone ethyl ester yield: 99%
N-carboxyethyl-noroxycodone ethyl ester yield: 98%
3.5. General Synthesis of N-carboxyethyl-nor-compounds
The specific N-carboxyethyl-nor-compound ethyl ester (0.5 mmol) was added to the mixture of ethanol (1 g) and water (2.5 mL). The mixture was reacted with sodium hydroxide (1 M, 0.5 mmol) for 1 h at 60 °C. After the reaction the pH was set to 3–4 with 10% HCl solution. Then the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
N-carboxyethyl-normorphine yield: 96%
N-carboxyethyl-dihydronormorphine yield: 96%
N-carboxyethyl-norcodeine yield: 93%
N-carboxyethyl-dihydronorcodeine yield: 95%
N-carboxyethyl-noroxymorphone yield: 99%
N-carboxyethyl-noroxycodone yield: 96%
3.6. General Synthesis of N-acetyl-glycine-nor-compound Ethyl Esters
The specific normorphine derivative (2 mmol) was dissolved in 30 mL of acetonitrile. In the presence of sodium bicarbonate (10 mmol) and potassium iodide (2.4 mmol) N-chloroacetyl-glycine ethyl ester (2.4 mmol) was added to the solution. The mixture was stirred and refluxed for 8 h. After the TLC showed no more starting compound the inorganic salts were filtered and the solvent was evaporated. If it was necessary column chromatography was used. (Choloroform: Methanol 9:1)
N-acetyl-glycine-normorphine ethyl ester yield: 94%
N-acetyl-glycine-dihydronormorphine ethyl ester yield: 91%
N-acetyl-glycine -norcodeine ethyl ester yield: 90%
N-acetyl-glycine -dihydronorcodeine ethyl ester yield: 89%
N-acetyl-glycine -noroxymorphone ethyl ester yield: 95%
N-acetyl-glycine -noroxycodone ethyl ester yield: 96%
3.7. General Synthesis of N-acetyl-glycine-nor-compounds
The specific N-acetyl-glycine-nor-compound ethyl ester (0.5 mmol) was added to the mixture of ethanol (1 g) and water (2.5 mL). The mixture was reacted with sodium hydroxide (1 M, 0.5 mmol) for 1 h at 60 °C. After the reaction the pH was set to 3–4 with 10% HCl solution. Then the solvent was evaporated to obtain the hydrochloride salt of the desired compound.
N-acetyl-glycine-normorphine yield: 98%
N-acetyl-glycine-dihydronormorphine yield: 94%
N-acetyl-glycine-norcodeine yield: 99%
N-acetyl-glycine-dihydronorcodeine yield: 95%
N-acetyl-glycine-noroxymorphone yield: 98%
N-acetyl-glycine-noroxycodone yield: 93%


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}