Synthesis and Characterization of Some 5-Acetylbarbituric Based Thiosemicarbazone Derivatives †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
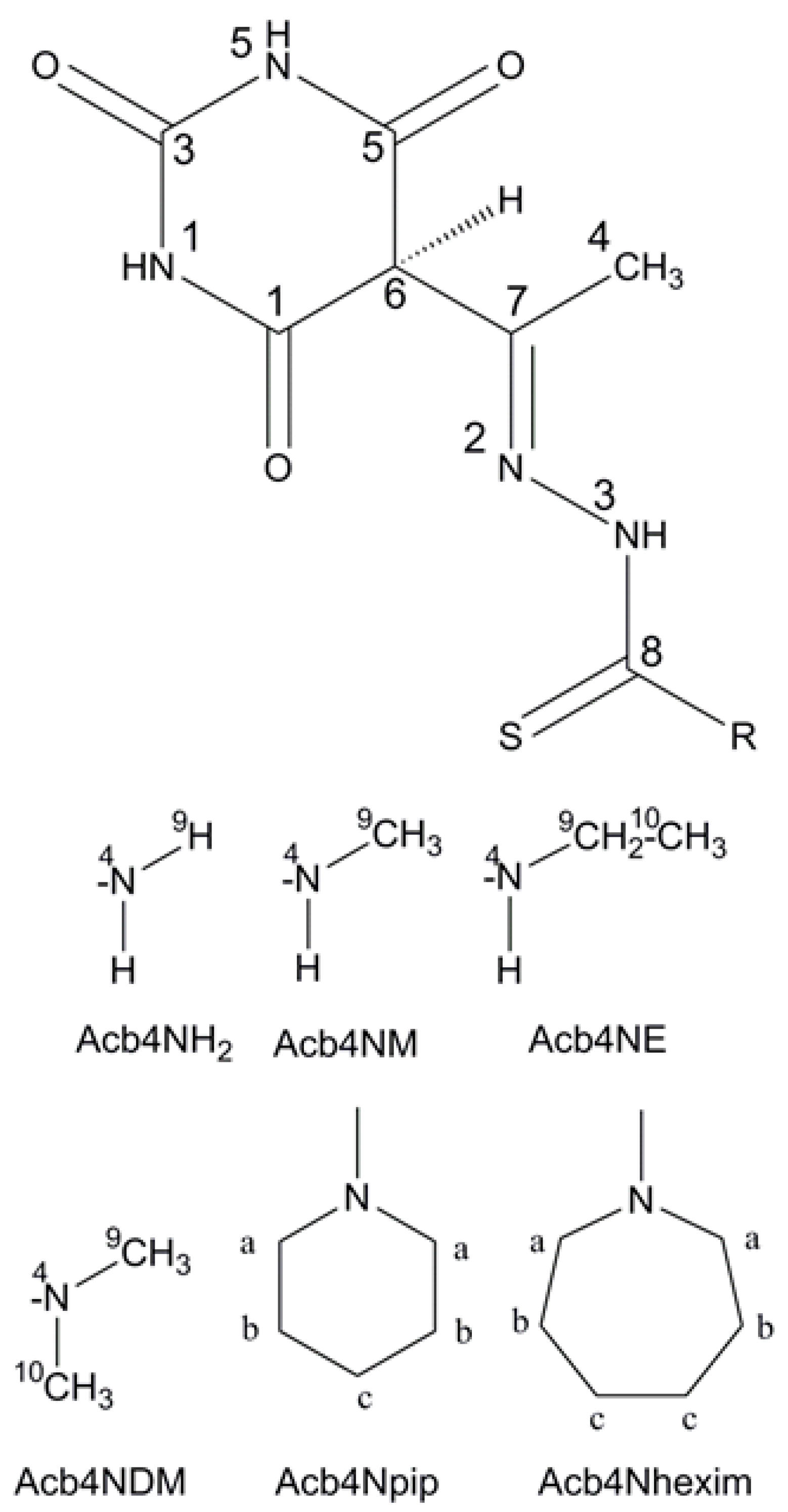
2. Materials and Methods
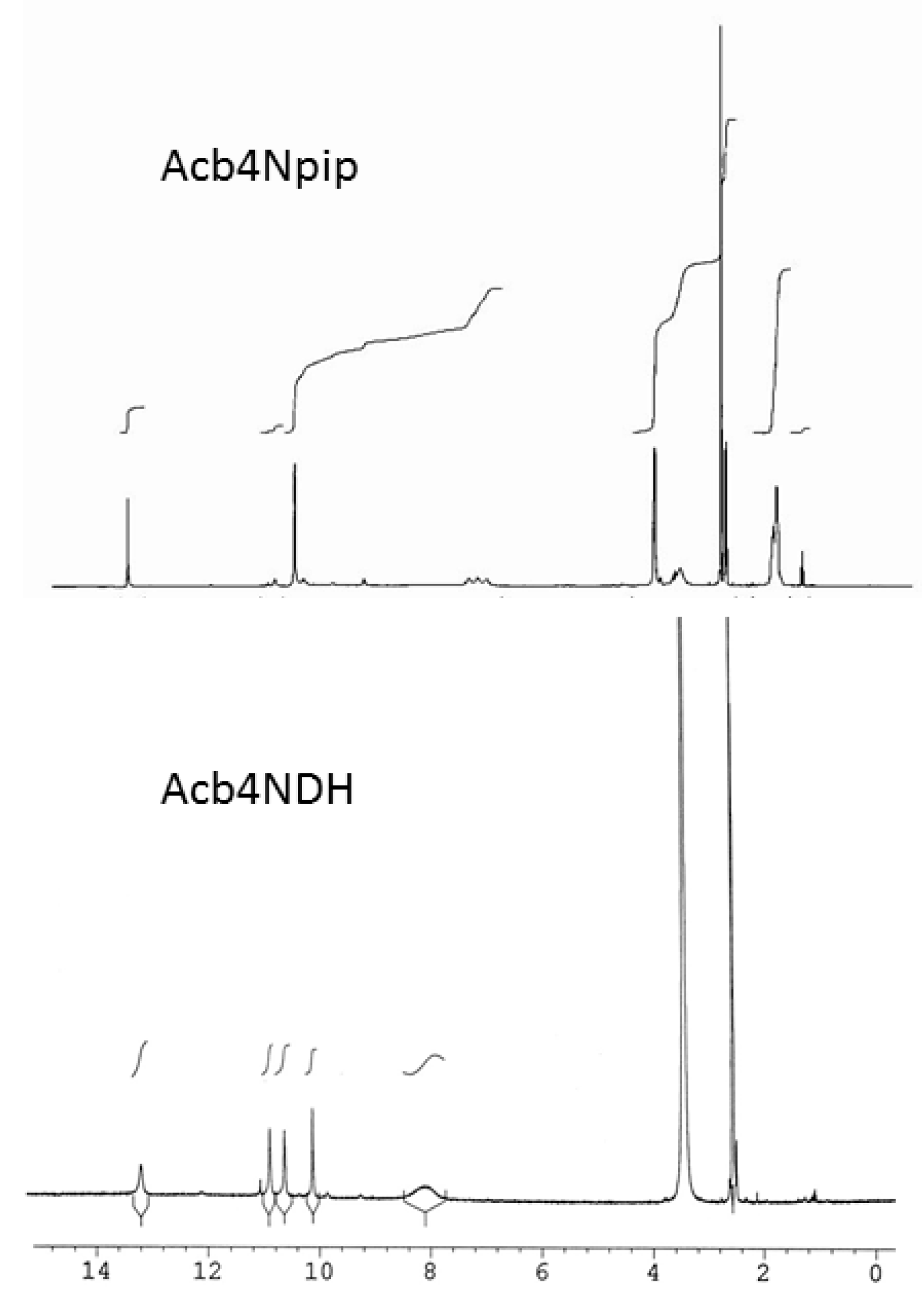
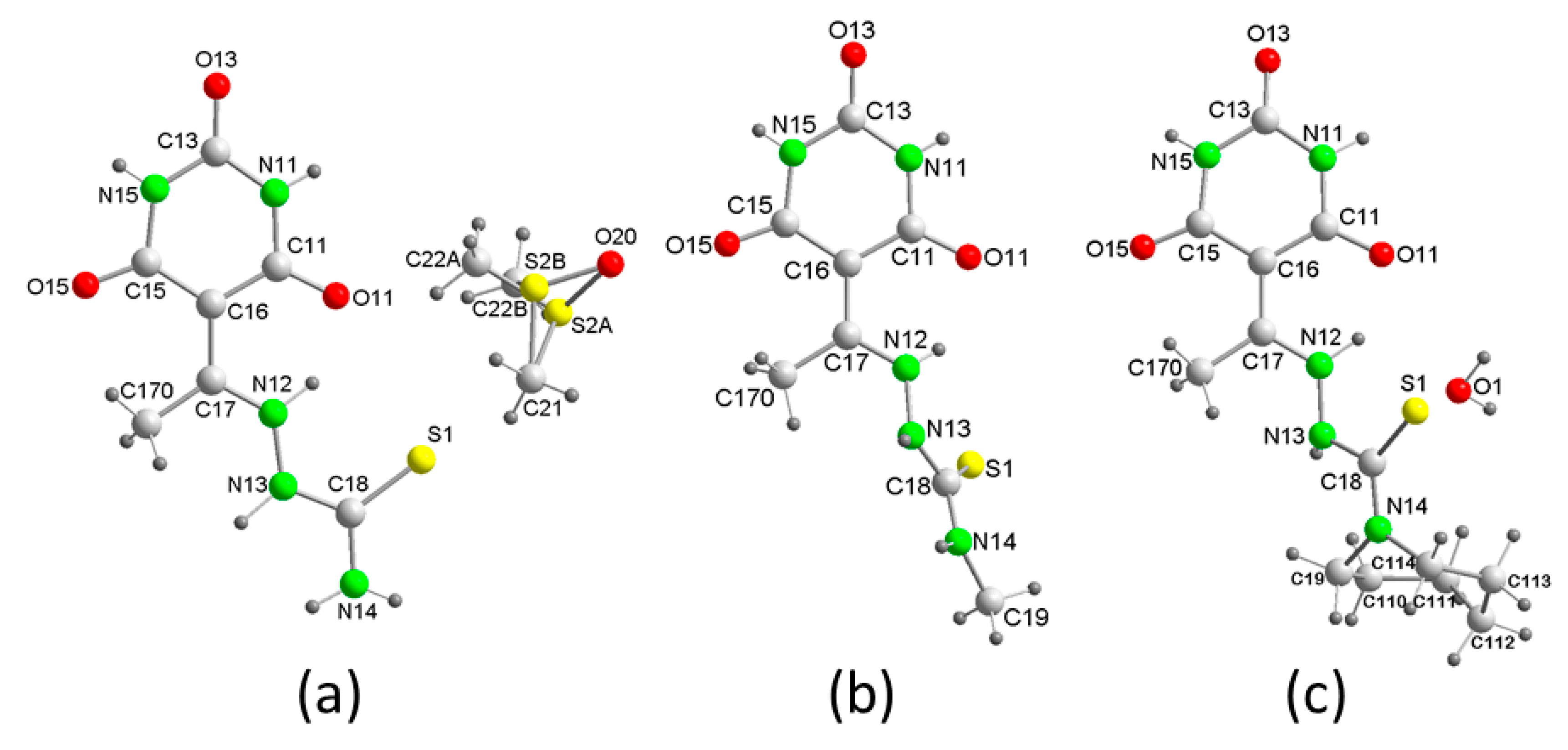
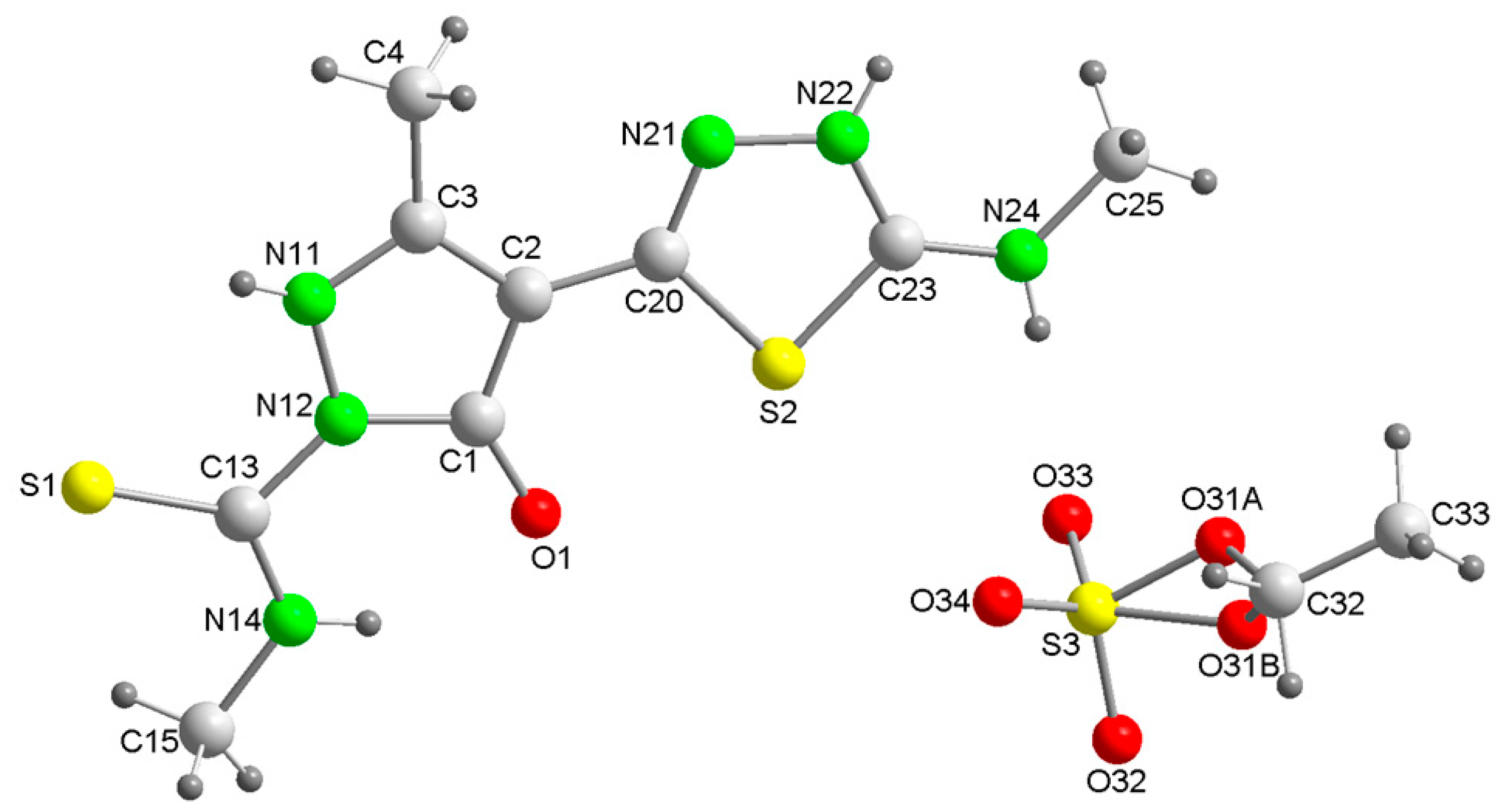
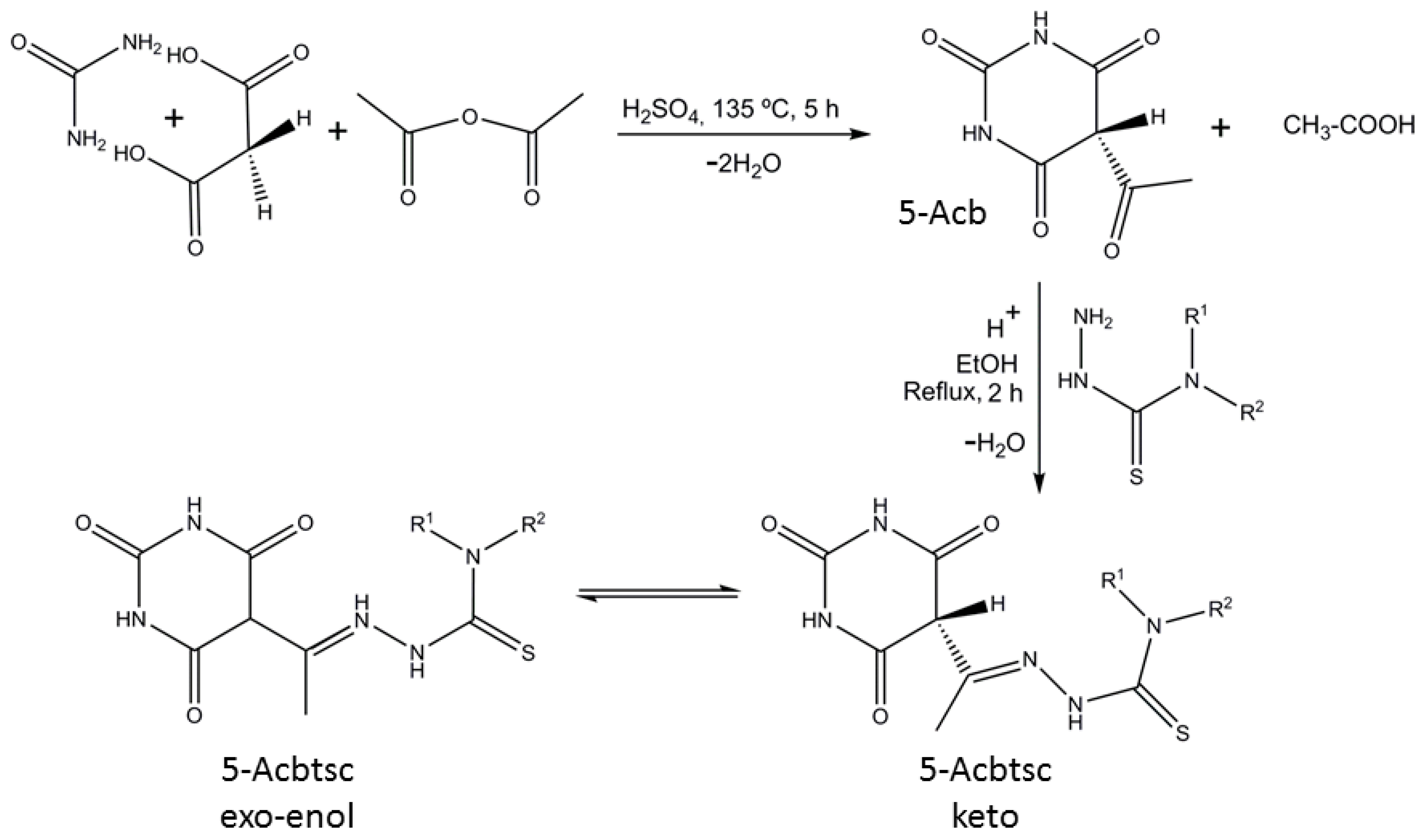
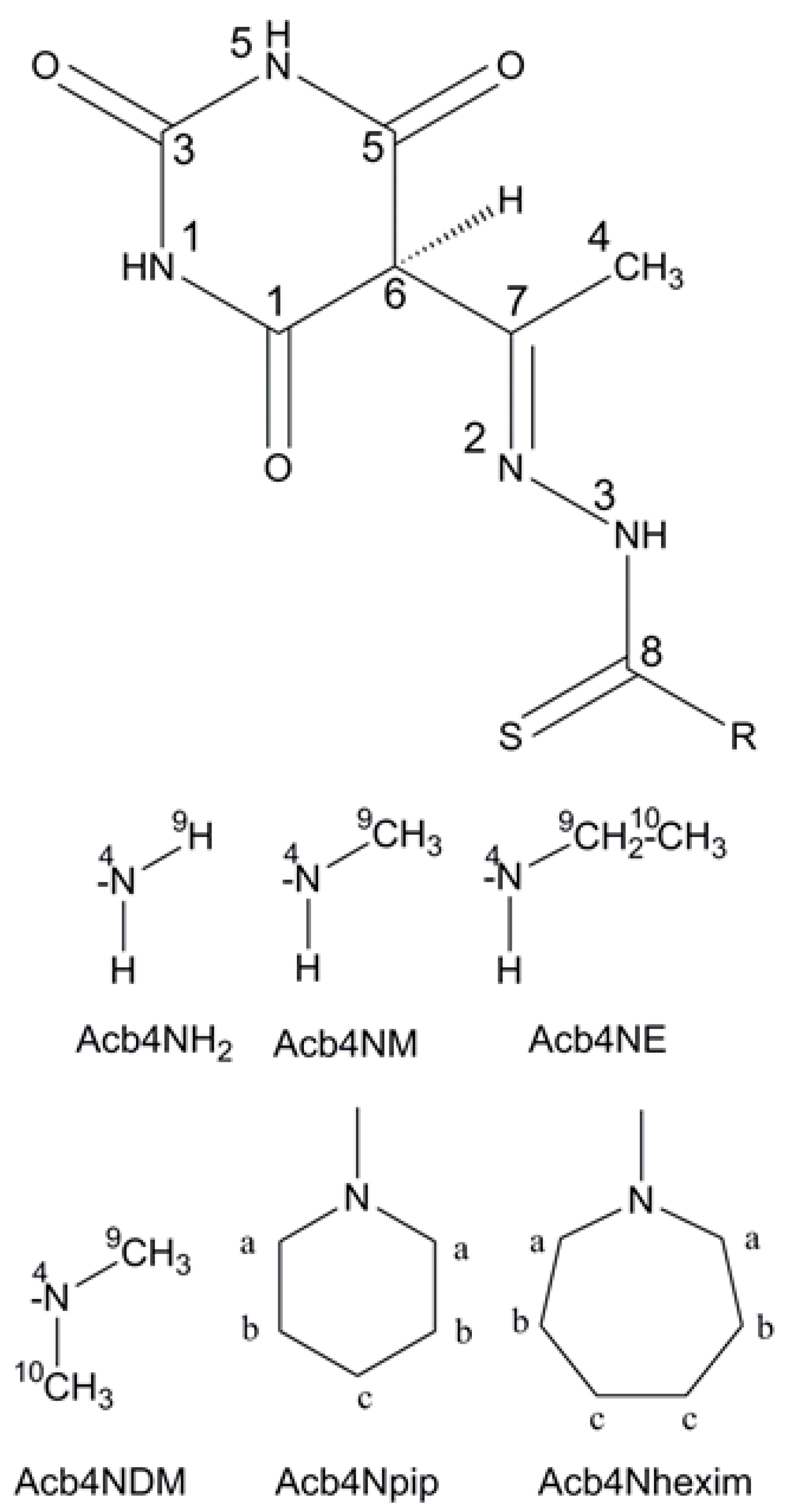
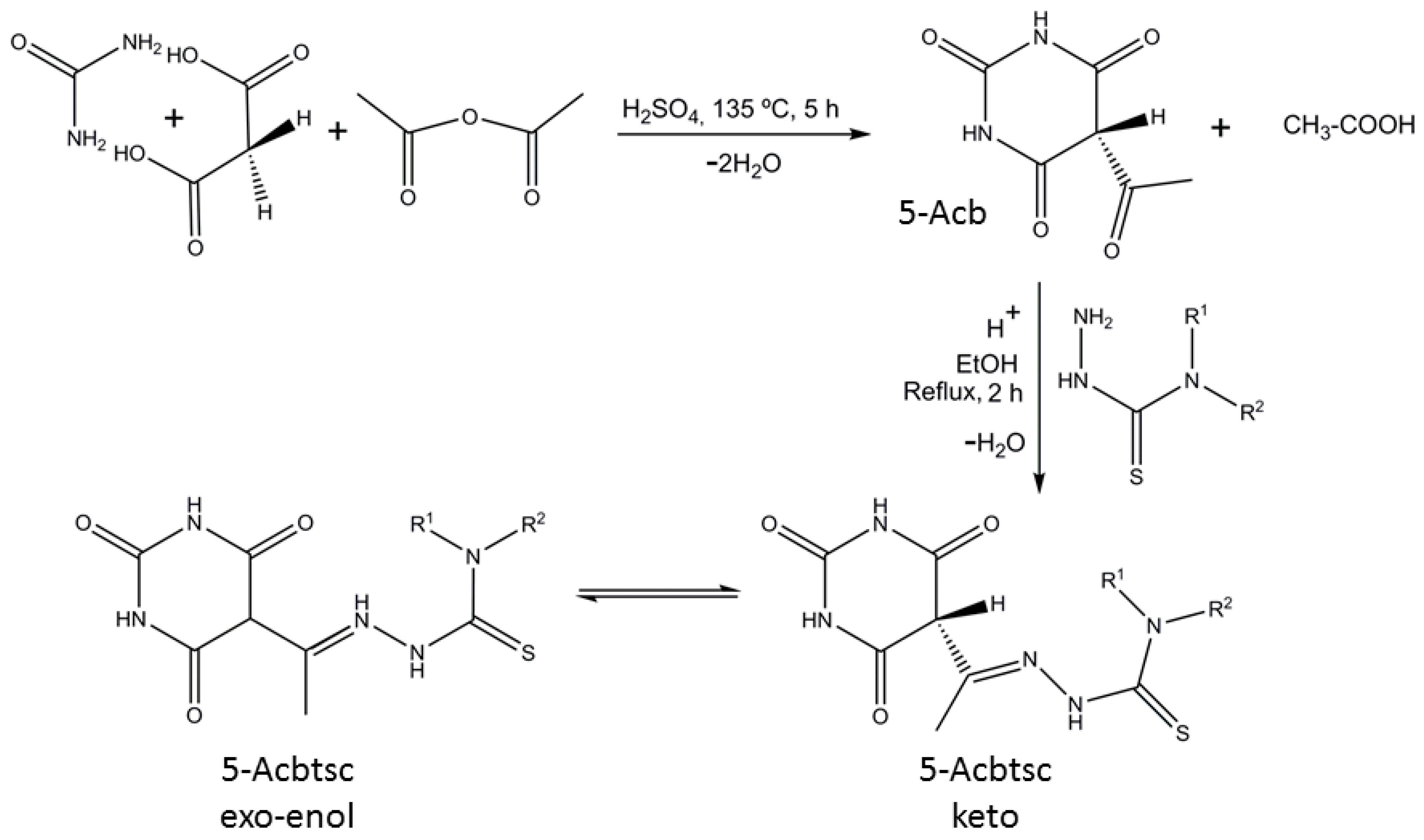
3. Results and Discussion
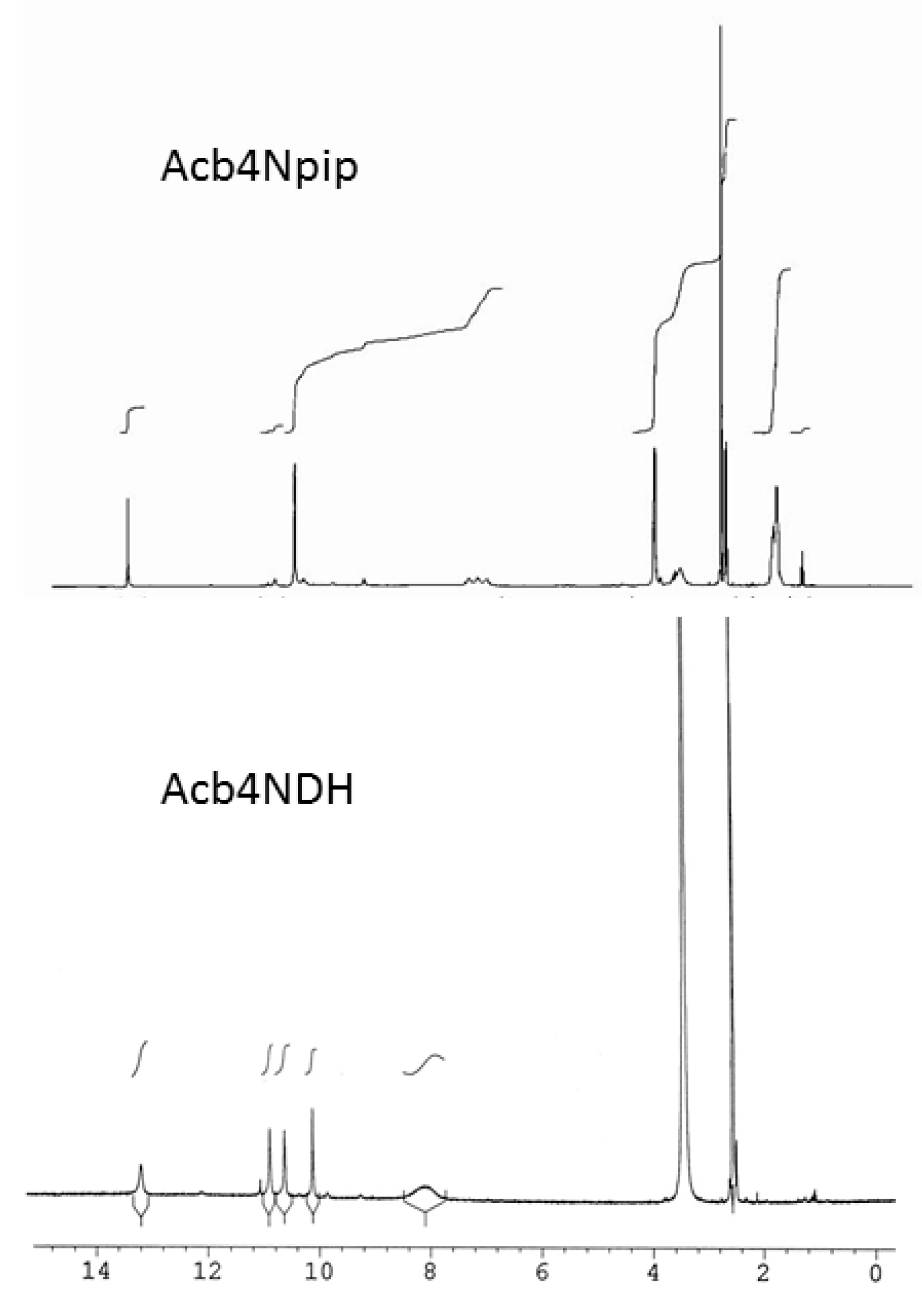
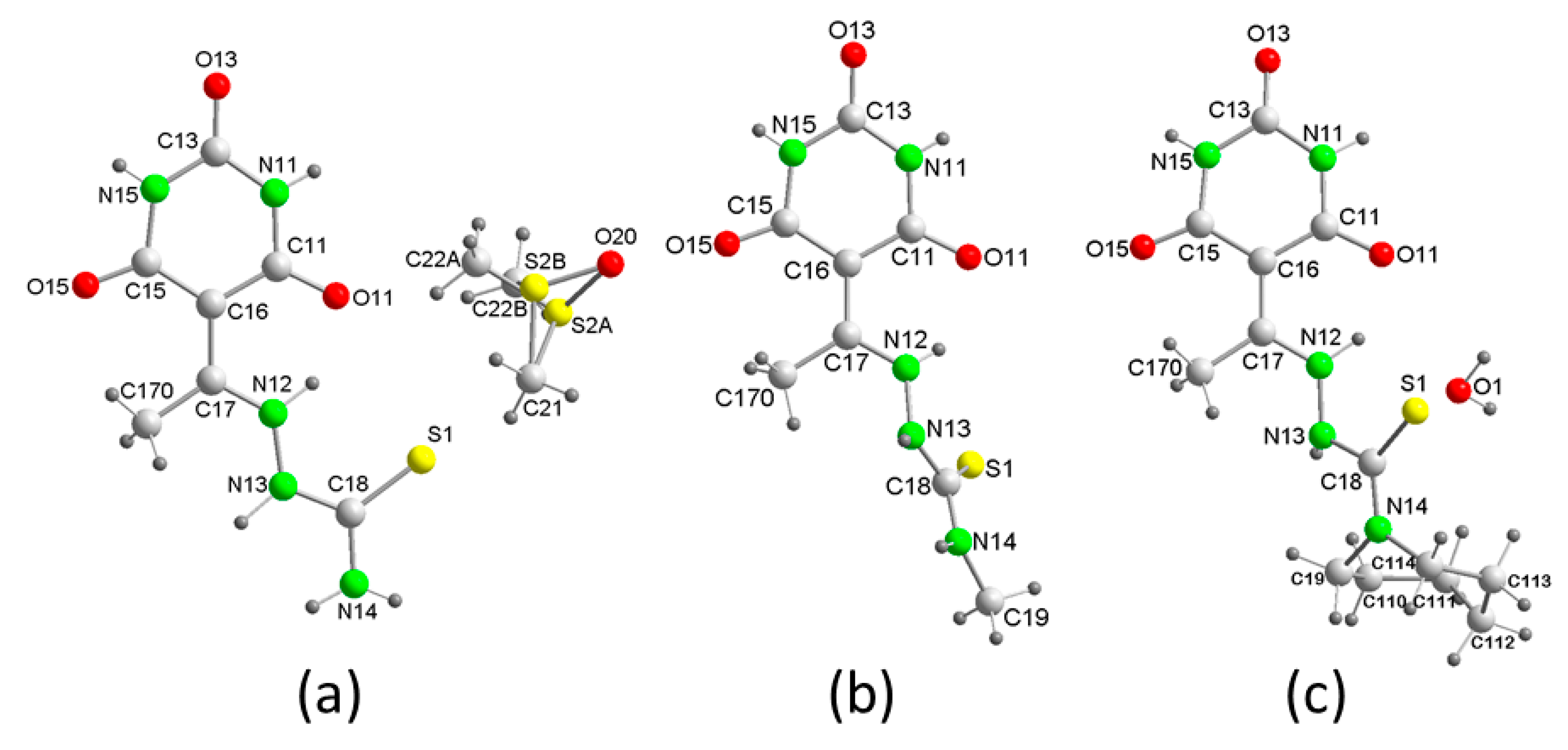
3.1. Structural
3.2. Antiproliferative Activity
Acknowledgments
Conflicts of Interest
References
- Castiñeiras, A.; García-Santos, I.; Nogueiras, S.; Rodríguez-González, I.; Rodríguez-Riobó, R. Supramolecular interactions in biologically relevant compounds. 2-Pyrazineformamide thiosemicarbazones and some products of their cyclization. J. Mol. Struct. 2014, 1074, 1–18. [Google Scholar] [CrossRef]
- Beraldo, H.; Gambino, D. The wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini-Rev. Med. Chem. 2004, 4, 31–39. [Google Scholar] [PubMed]
- Paterson, B.M.; Donnelly, P.S. Copper complexes of bis(thiosemicarbazones): From chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem. Soc. Rev. 2011, 40, 3005–3018. [Google Scholar] [CrossRef] [PubMed]
- van Koningsbruggen, P.J.; Haasnoot, J.G.; de Graaff, R.A.G.; Reedijk, J. Syntheses, spectroscopic and magnetic properties and X-ray crystal structure of two dinuclear copper(II) compounds with the ligand Na-salicyloylpyridine-2-carboxamidrazonato. Inorg. Chim. Acta 1995, 235, 87–94. [Google Scholar] [CrossRef]
- Sharma, A.; Jad, Y.; Siddiqui, M.R.H.; de la Torre, B.G.; Albericio, F.; El-Faham, A. Synthesis, Characterization, and Tautomerism of 1,3-Dimethyl Pyrimidine-2,4,6-Trione s-Triazinyl Hydrazine/Hydrazone Derivatives. J. Chem. 2017, 2017, 5702962. [Google Scholar] [CrossRef]
- Bermejo, E.; Castiñeiras, A.; García-Santos, I.; Gómez-Rodríguez, L.; Sevillano, P. Pt4S4 Clusters with Functionalized Thiosemicarbazones derived from 5-Acetylbarbituric Acid. Z. Anorg. Allg. Chem. 2007, 633, 2255–2261. [Google Scholar] [CrossRef]
- Biltz, H.; Wittek, H. Über alkylierte und acylierte barbitursaüren. Chem. Ber. 1921, 54, 1035–1058. [Google Scholar] [CrossRef]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Lim Xue Ting, R.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; et al. New Water-Soluble Copper(II) Complexes with Morpholine−Thiosemicarbazone Hybrids: Insights into the Anticancer and Antibacterial Mode of Action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiñeiras, A.; Fernández-Hermida, N.; García-Santos, I.; Gómez-Rodríguez, L. Synthesis and Characterization of Some 5-Acetylbarbituric Based Thiosemicarbazone Derivatives. Proceedings 2019, 41, 10. https://doi.org/10.3390/ecsoc-23-06479
Castiñeiras A, Fernández-Hermida N, García-Santos I, Gómez-Rodríguez L. Synthesis and Characterization of Some 5-Acetylbarbituric Based Thiosemicarbazone Derivatives. Proceedings. 2019; 41(1):10. https://doi.org/10.3390/ecsoc-23-06479
Chicago/Turabian StyleCastiñeiras, Alfonso, Nuria Fernández-Hermida, Isabel García-Santos, and Lourdes Gómez-Rodríguez. 2019. "Synthesis and Characterization of Some 5-Acetylbarbituric Based Thiosemicarbazone Derivatives" Proceedings 41, no. 1: 10. https://doi.org/10.3390/ecsoc-23-06479
APA StyleCastiñeiras, A., Fernández-Hermida, N., García-Santos, I., & Gómez-Rodríguez, L. (2019). Synthesis and Characterization of Some 5-Acetylbarbituric Based Thiosemicarbazone Derivatives. Proceedings, 41(1), 10. https://doi.org/10.3390/ecsoc-23-06479