π-π Stacking Interactions of 3a-Aryl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-ones, X-Ray and DFT Study †



Abstract
:1. Introduction
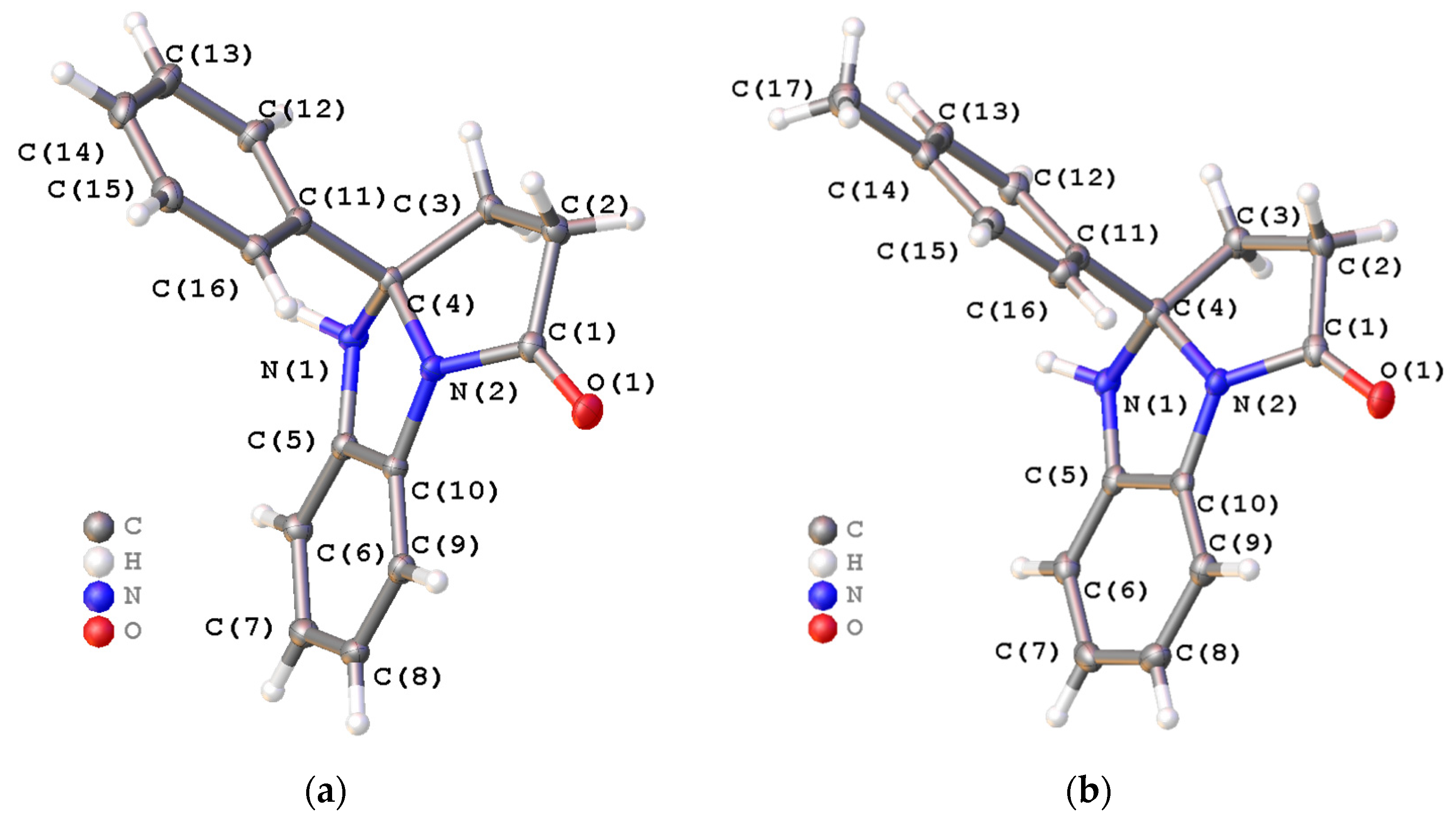
2. Results
3. Discussion
4. Materials and Methods
4.1. Synthesis of the Compounds
4.2. X-ray Study
4.3. DFT Study
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| CCDC | The Cambridge Crystallographic Data Centre |
| PD | Parallel displaced |
| RFBR | Russian Foundation for Basic Research |
| SE | Electrophilic substitution reactions |
References
- Fahlman, B.D. Solid-state chemistry. In Materials Chemistry; Springer: Dordrecht, The Netherlands, 2007; Chapter 2; pp. 13–85. [Google Scholar]
- Li, L.; Zhu, M.-L.; Lu, L.-P. (Glycylglycinato-[kappa]3O,N,N′)(2-methyl-1H-benzimidazole-[kappa]N3)copper(II) trihydrate. Acta Cryst. Sect. C 2006, 62, m227–m228. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, S.-L.; Zheng, S.-R.; Zhang, W.-G. Two mononuclear octahedral complexes with benzimidazole-2-carboxylate: Supramolecular networks constructed by hydrogen bonds. Acta Cryst. Sect. C 2011, 67, m346–m350. [Google Scholar] [CrossRef] [PubMed]
- Grinev, V.S.; Egorova, A.Y. 3a-Phenyl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-one, a potential plant-growth regulator. Acta Cryst. Sect. C 2013, 69, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Grinev, V.S.; Lyubun’, E.V.; Egorova, A.Y. Agrokhimiya (Agrochemistry) 2011, 3, 46–50. (In Russian)
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GAUSSIAN09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]



{kind=link}
{kind=link}
| Compd | D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|---|
| 1a | N(1)-H(1N)...O(1) #1 | 0.914(16) | 2.112(17) | 2.9994(11) | 163.4(14) |
| 1b | N(1)-H(1N)...O(1) #2 | 0.88(2) | 2.20(2) | 3.0509(17) | 161.2(2) |
| Compd | X-Ray | MPWB95/ 6-31G(d) | MPWB95/ 6-31++G | MPWB95/ 6-31++G(d) | M06-2X/ 6-31G(d) | M06-2X/ 6-31++G | M06-2X/ 6-31++G(d) |
|---|---|---|---|---|---|---|---|
| 1a | 4.258 | 4.284 1 | 4.285 | 4.276 | 3.938 | 3.970 | 3.950 |
| −4.37 2 | −4.15 | −3.81 | −10.42 | −10.77 | −10.58 | ||
| 1b | 4.386 | 5.246 | 4.678 | 5.448 | 4.090 | 4.094 | 4.167 |
| −4.40 | −3.81 | −3.25 | −9.25 | −9.88 | −9.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinev, V.; Yegorova, A. π-π Stacking Interactions of 3a-Aryl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-ones, X-Ray and DFT Study. Proceedings 2018, 2, 1120. https://doi.org/10.3390/IECC_2018-05255
Grinev V, Yegorova A. π-π Stacking Interactions of 3a-Aryl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-ones, X-Ray and DFT Study. Proceedings. 2018; 2(14):1120. https://doi.org/10.3390/IECC_2018-05255
Chicago/Turabian StyleGrinev, Vyacheslav, and Alevtina Yegorova. 2018. "π-π Stacking Interactions of 3a-Aryl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-ones, X-Ray and DFT Study" Proceedings 2, no. 14: 1120. https://doi.org/10.3390/IECC_2018-05255
APA StyleGrinev, V., & Yegorova, A. (2018). π-π Stacking Interactions of 3a-Aryl-2,3,3a,4-tetrahydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-1-ones, X-Ray and DFT Study. Proceedings, 2(14), 1120. https://doi.org/10.3390/IECC_2018-05255