1. Introduction
In early 1990s, there were two works published with the description of two different polymorphs of 4-oxo-4-phenylbutanoic acid, which were deposited in CCDC with codes VERMAG and VERMAG01, respectively [
1,
2]. The crystal of the first one, VERMAG, was obtained from methanol, while the second modification was synthesized in benzene. Both polymorphs crystallize in monoclinic space groups, P2
1/c (VERMAG) and the non-standard space group P2
1/n (VERMAG01), and have similar parameters of the crystal lattice.
Today, scientists pay great attention to crystals with Z′ ≥ 2. Reported here new polymorph of 4-oxo-4-phenylbutanoic acid crystallizes also in P21/c space group, but the parameters of crystal lattice differ from published before crystals significantly.
2. Results
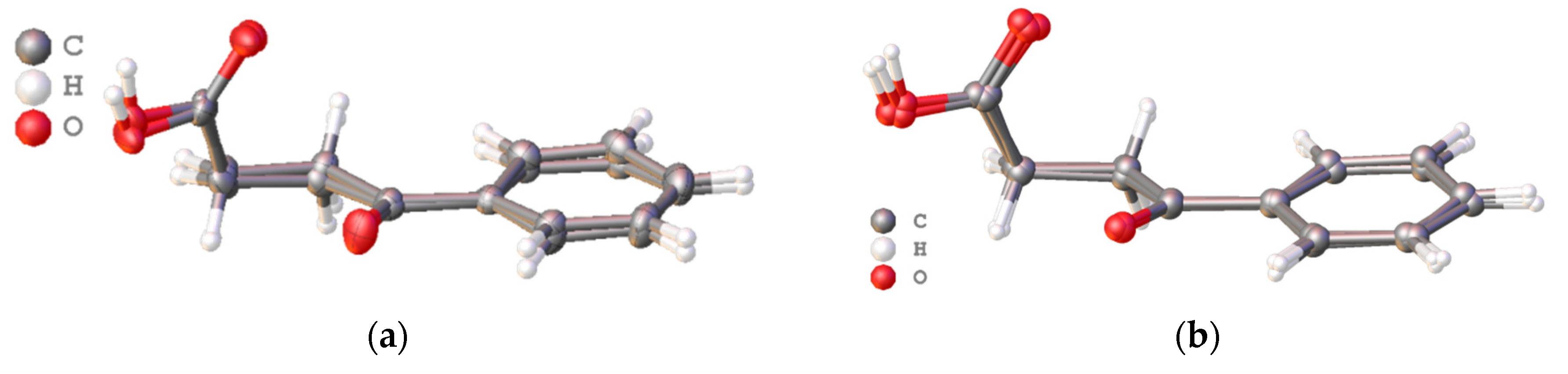
Structurally, polymorphic modification of 4-oxo-4-phenylbutanoic acid differs from deposited in CCDC with codes VERMAG and VERMAG01 crystals very slightly (RMSD of about 0.112–0.183 Å). The asymmetrical unit of reported crystal contains four pairs of crystallographically independent molecules (
Z = 8,
Z′ = 2) with RMSD of 0.241 Å (with inversion) (
Figure 1a). It is interesting that, according to the RMSD values, two crystallographically independent molecules in reported here crystal differ from each other more significantly than the molecules from all known polymorphs (
Figure 1b).
All polymorphs contain dimers of molecules bounded by intermolecular hydrogen bonds between carboxyl groups. No interactions observed with oxygen atom of carbonyl group at position 4. The summarized H-bonds data for all considering crystals given in
Table 1. It can be seen that the H…O distances in our crystal differ only by 0.036 Å, while the differences between this structure and VERMAG as well as VERMAG01 significantly more, and are 0.179 and 0.108 Å, correspondingly. It is remarkable that the H…O distance of studied crystal of 4-oxo-4-phenylbutanoic acid lies in between VERMAG and VERMAG01. The D…A distances of two molecules in studied crystal as well as in VERMAG01 polymorph are very similar (about 2.65 Å), in contrast with the VERMAG where this distance is about 0.1 Å longer.
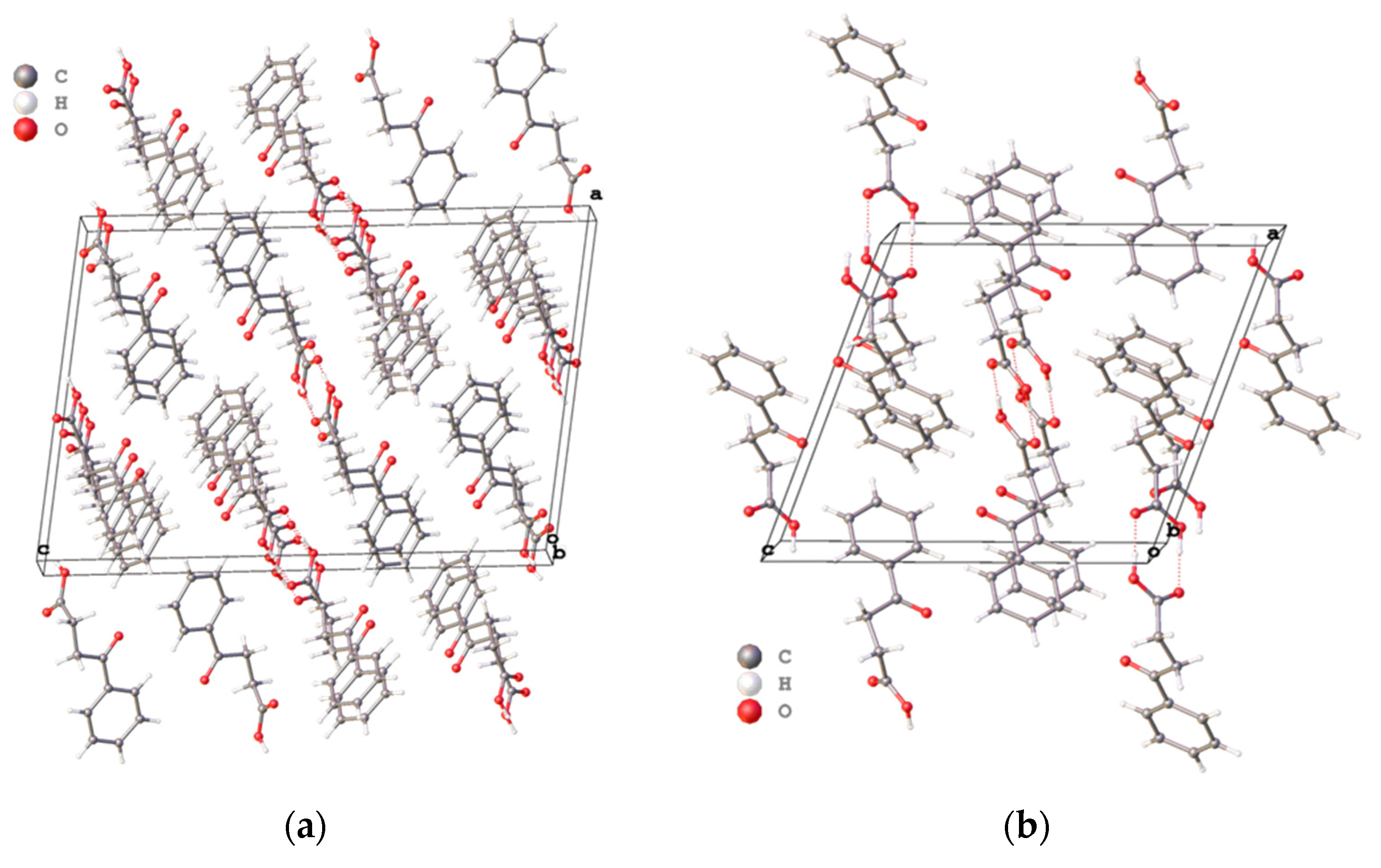
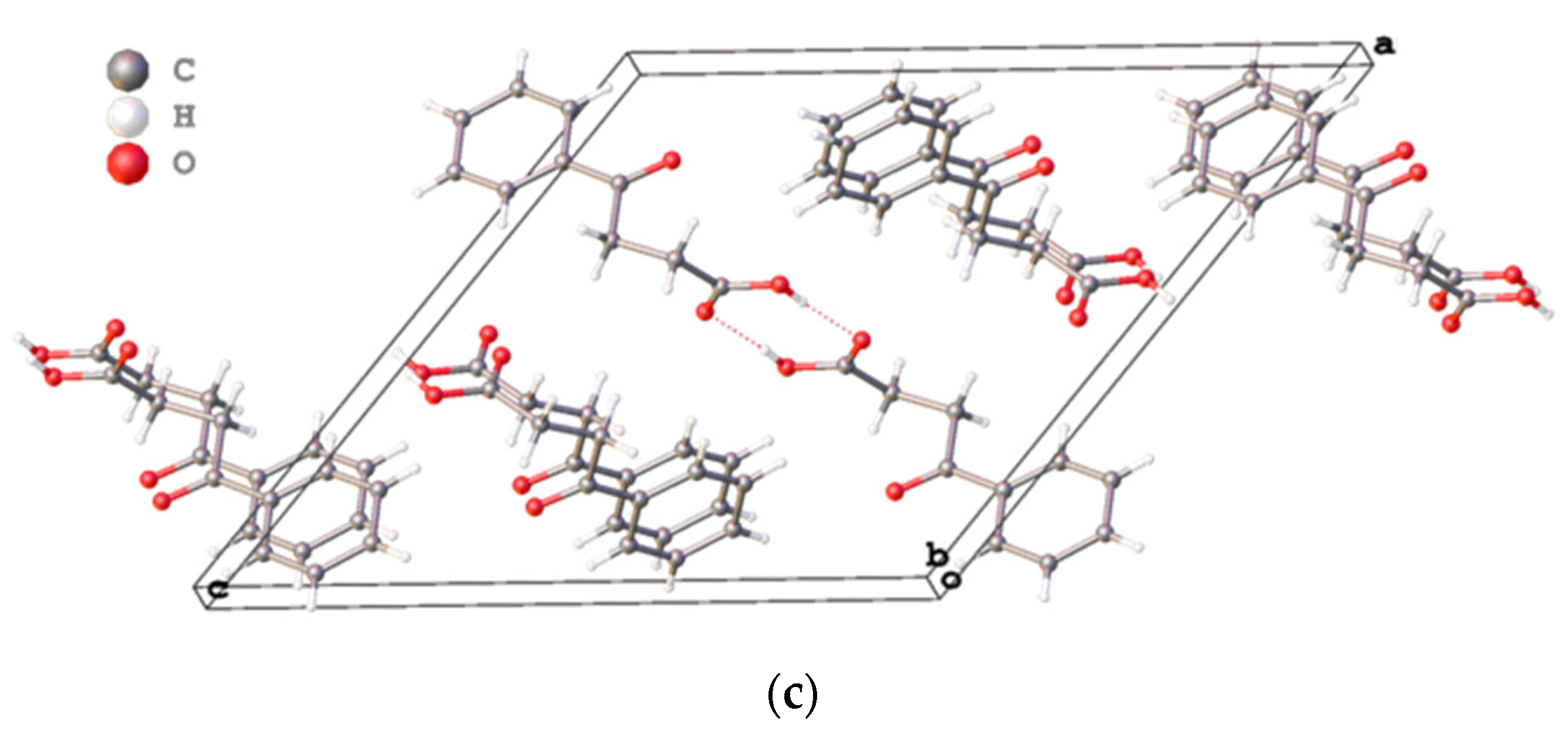
Main differences of three polymorphs are in the crystal packing (
Figure 2a–c). Briefly, the crystal package of VERMAG with the volume cell of 1013.91 Å
3 is the least dense of all crystals, while the reported polymorph has the most dense packing with the volume cell of 1754.51 Å
3 and two sets of molecules in comparison to early known modifications.
3. Discussion
The reported monoclinic crystal of 4-oxo-4-phenylbutanoic acid belongs to the space group P21/c and has significant larger cell volume. Selladurai et al. reported their structure (VERMAG) as a monoclinic crystal of the space group P21/c with an β angle of 129.57(10)°, while Thompson et al. refine their structure (VERMAG01) in the space group P21/n with the less-obtuse β angle of 111.33(3)°. To the best of our knowledge, more accurate results can be achieved when use P21/c for angles of less than 120°. So, our structure was refined in the space group P21/c with more sharp β angle of 98.0217(7)°.
It was surprising that, structurally, two crystallographically independent molecules in reported crystal differ each other more than the molecules from all known polymorphs. This can be explained by significant flexibility of the alkyl units of the acid.
Considering all three known crystals of 4-oxo-4-phenylbutanoic acid, our polymorph has the most dense package. In all crystals molecules of the acid are formed dimers using carboxyl groups which is in harmony with the common data for all carboxylic acids. The parameters of intermolecular hydrogen bonds of our crystal lies between corresponding values of known polymorphs and looks ordinary.
4. Materials and Methods
4-oxo-4-phenylbutanoic acid was synthesized by Friedel–Crafts condensation from succinic anhydride and benzene with anhydrous AlCl3 catalysis, recrystallized from water and dried under vacuum. M.p. 112–114 °C. The single crystal for X-ray study was obtained from benzene by slow evaporation.
X-ray diffraction was performed on an automatic three-circle diffractometer Bruker SMART Apex II (graphite monochromator, λ(MoKα) = 0.71073 Å, ω scan) at 120 K. Integration of intensities was carried out using the procedure built into the software complex SAINT [
3]. Semi-empirical corrections for absorption and for systematic errors are based on the intensity of equivalent reflections in the program SADABS [
4]. The structure was solved by a direct method and was refined by full-matrix least-squares versus
F2hkl with anisotropic displacement parameters for all non-hydrogen atoms. The hydrogen atoms of the OH groups were found from the difference Fourier series and refined in the isotropic approximation. The other hydrogen atoms were placed in the calculated positions and were refined geometrically by using a riding model with
Uiso(H) = 1.2
Uiso(C) and
Uiso(H) = 1.5
Uiso(C) for methyl and other groups. Solving and refinement were carried out using the SHELX software package version 2016/6 [
5]. The overlays and packing diagrams as well as parameters of non-covalent interactions were obtained using Olex2 software [
6].
Author Contributions
E.L. and E.K. carried out the synthesis of reported compound and obtained the single crystal, V.G. took part in the spectral characterization of synthesized compound, drafted the manuscript, prepared final version of the manuscript, and assisted technically, A.Y. designed and supervised all experiments, and participated in manuscript drafting. All authors read and approved the final version of the manuscript.
Acknowledgments
The work was supported by the RFBR (research project no. 16-03-00530).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| CCDC | The Cambridge Crystallographic Data Centre |
| RFBR | Russian Foundation for Basic Research |
References
- Selladurai, S.; Kumar, M.S.; Subramanian, K. Crystal and molecular structure of 3-benzoylpropionic acid. Proc. Indian Acad. Sci. Chem. Sci. 1990, 102, 39–43. [Google Scholar] [CrossRef]
- Thompson, H.W.; Vanderhoff, P.A.; Lalancette, R.A. 3-Benzoylpropionic acid: structure and hydrogen-bonding pattern. Acta Cryst. Sect. C 1991, 47, 1443–1445. [Google Scholar] [CrossRef]
- Bruker. SAINT v7.23A; Bruker AXS Inc.: Billerica, MA, USA, 2005. [Google Scholar]
- Sheldrick, G.M. SADABS v2008/1, Bruker/Siemens Area Detector Absorption Correction Program; Bruker AXS Inc.: Billerica, MA, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




{kind=link}
{kind=link}
{kind=link}