
The First Reaction Steps of Lithium-Mediated Ammonia Synthesis: Ab Initio Simulation


Abstract
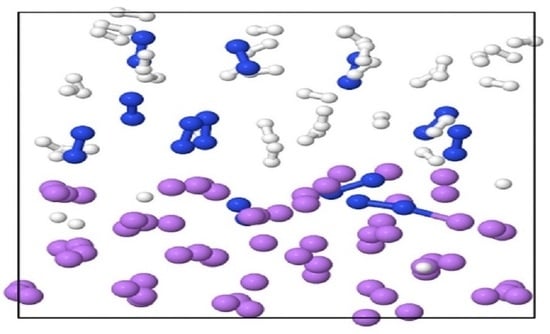
:
1. Introduction
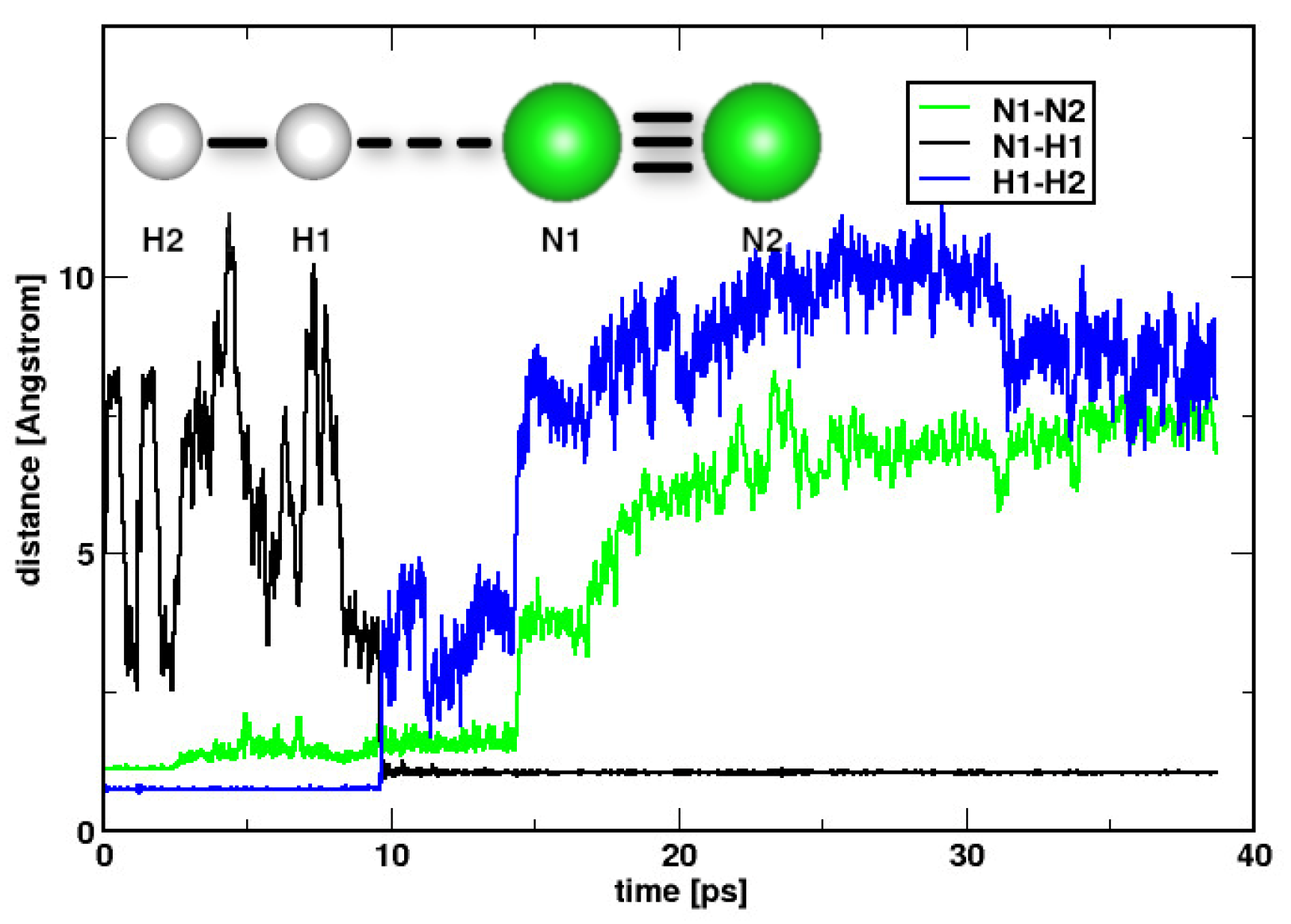
2. Results and Discussions
3. Conclusions
4. Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIMD | Ab-Initio Molecular Dynamics |
| BLYP | Becke-Lee-Yang-Parr density functional |
| B3LYP | Becke-Lee-Yang-Parr hybrid density functional with three parameters |
| B2PLYP | Double hybrid functional: B3LYP combined with 2nd order perturbation theory |
| CCSD | Coupled-Cluster, singles, doubles |
| CPMD | Car-Parrinello Molecular Dynamics |
| D3BJ | Dispersion correction 3, Becke-Johnson damping |
| DFT | Density functional theory |
| MP2 | Moller-Plesset perturbation theory, 2nd order |
References
- Holleman, A.F.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 101st ed.; de Gruyter: Berlin, Germany; New York, NY, USA, 1995. [Google Scholar]
- Wikipedia. Available online: https://en.wikipedia.org/wiki/Ammonia–production (accessed on 16 June 2022).
- NIST Chemistry WebBook. Available online: https://webbook.nist.gov/ (accessed on 16 June 2022).
- Christensen, C.H.; Johannessen, T.; Sørensen, R.Z.; Nørskov, J.K. Towards an ammonia-mediated hydrogen economy? Catal. Today 2006, 111, 140. [Google Scholar] [CrossRef]
- Giddey, S.; Badwal, S.P.S.; Kulkami, A. Review of electrochemical ammonia production tech- nologies and materials. Int. J. Hydrogen Energy 2013, 38, 14576. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Choi, J.; Suryanto, B.H.R.; Jalili, R.; Chatti, M.; Azofra, L.M.; Simonov, A.N. Liquefied sunshine: Transforming renewables into fertilizers and energy carriers with electromaterials. Adv. Mater. 2020, 32, e1904804. [Google Scholar] [CrossRef] [PubMed]
- Tsuneto, A.; Kudo, A.; Sakata, T. Efficient electrochemical reduction of N2 to NH3 catalyzed by lithium. Chem. Lett. 1993, 22, 851. [Google Scholar] [CrossRef]
- Tsuneto, A.; Kudo, A.; Sakata, T. Lithium-mediated electrochemical reduction of high pressure N2 to NH3. J. Electroanalytical Chem. 1994, 367, 183. [Google Scholar] [CrossRef]
- Lazouski, N.; Schiffer, Z.J.; Williams, K.; Manthiram, K. Understanding Continuous Lithium- Mediated Electrochemical Nitrogen Reduction. Joule 2019, 3, 1127. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Andersen, S.Z.; Statt, M.J.; Saccoccio, M.; Bukas, V.J.; Krempl, K.; Sazinas, R.; Pedersen, J.B.; Shadravan, V.; Zhou, Y.; et al. Enhancement of lithium-mediated ammonia synthesis by addtion of oxygen. Science 2021, 374, 1593. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Langel, W.; Parrinello, M. Hydrolysis at stepped MgO surfaces. Phys. Rev. Lett. 1994, 73, 504. [Google Scholar] [CrossRef]
- Langel, W. Car-Parrinello simulation of H2O dissociation on rutile. Surf. Sci. 2002, 496, 141. [Google Scholar] [CrossRef]
- Tachikawa, H. Mechanism of Dissolution of a Lithium Salt in an Electrolytic Solvent in a Lithium Ion Secondary Battery: A Direct Ab Initio Molecular Dynamics (AIMD) Study. ChemPhysChem 2014, 15, 1604. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, E.; Brown, M.A.; Sterrer, M.; Freund, H.H.; Kwapien, K.; Sierka, M.; Sauer, J. Thickness-dependent hydroxylation of MgO(001) thin films. J. Phys. Chem. C 2010, 114, 18207. [Google Scholar] [CrossRef]
- Kirchner, B.; Seitsonen, A.P.; Hutter, J. CPMD, Version 4.1, Copyright IBM Corp 1990–2015, Copyright MPI für Festkörperforschung Stuttgart 1997–2001. Available online: http://www.cpmd.org/ (accessed on 25 May 2022).
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Boero, M.; Parrinello, M.; Terakura, K.; Weiss, H. Car-Parrinello study of Ziegler-Natta heterogeneous catalysis: Stability and destabilization problems of the active site models. Mol. Phys. 2002, 100, 2935–2940. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A simple effective potential for exchange. J. Chem. Phys. 2006, 124, 221101. [Google Scholar] [CrossRef]
- Becke, A. A New Mixing of Hartree-Fock and Local-Density Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
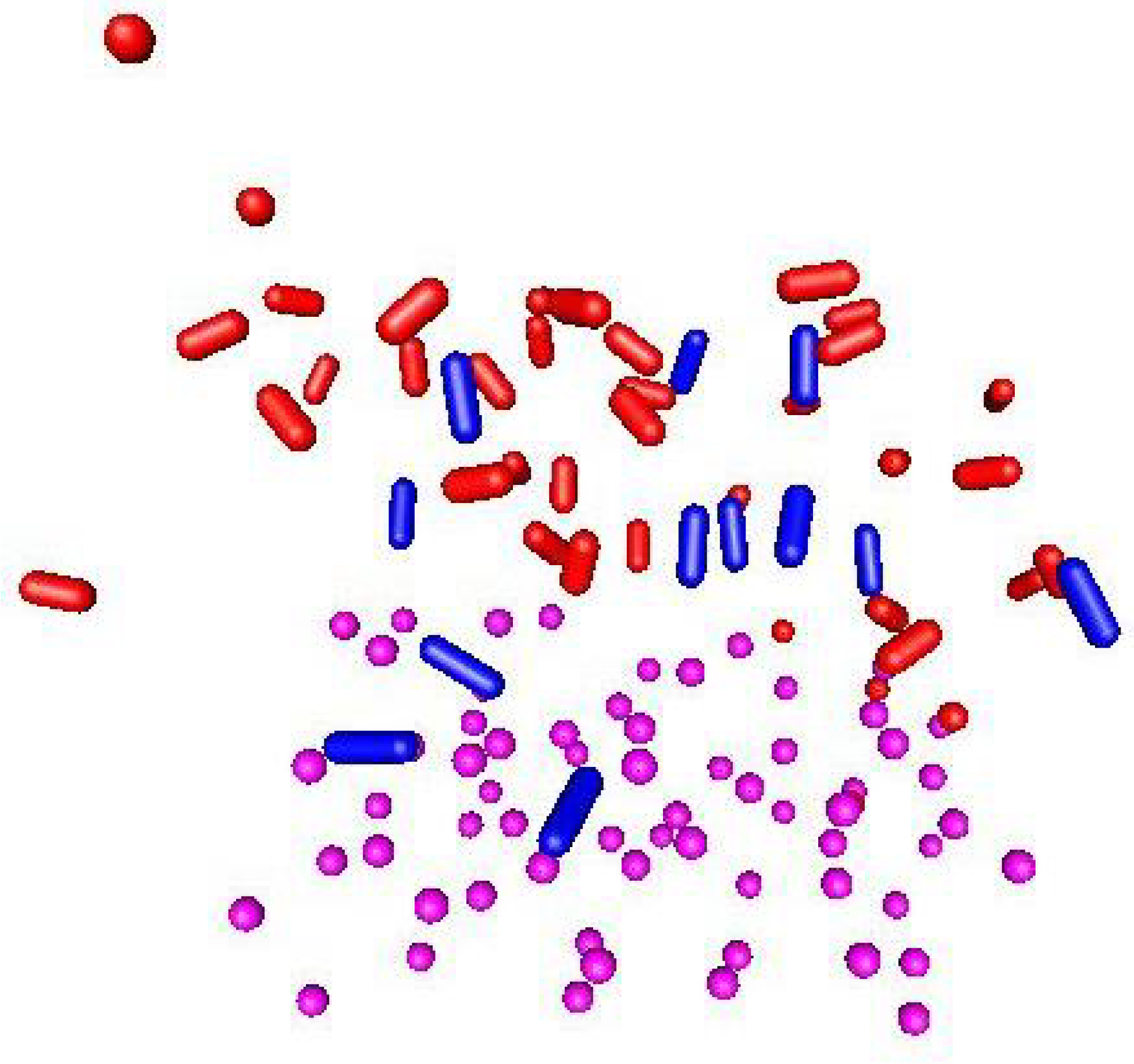

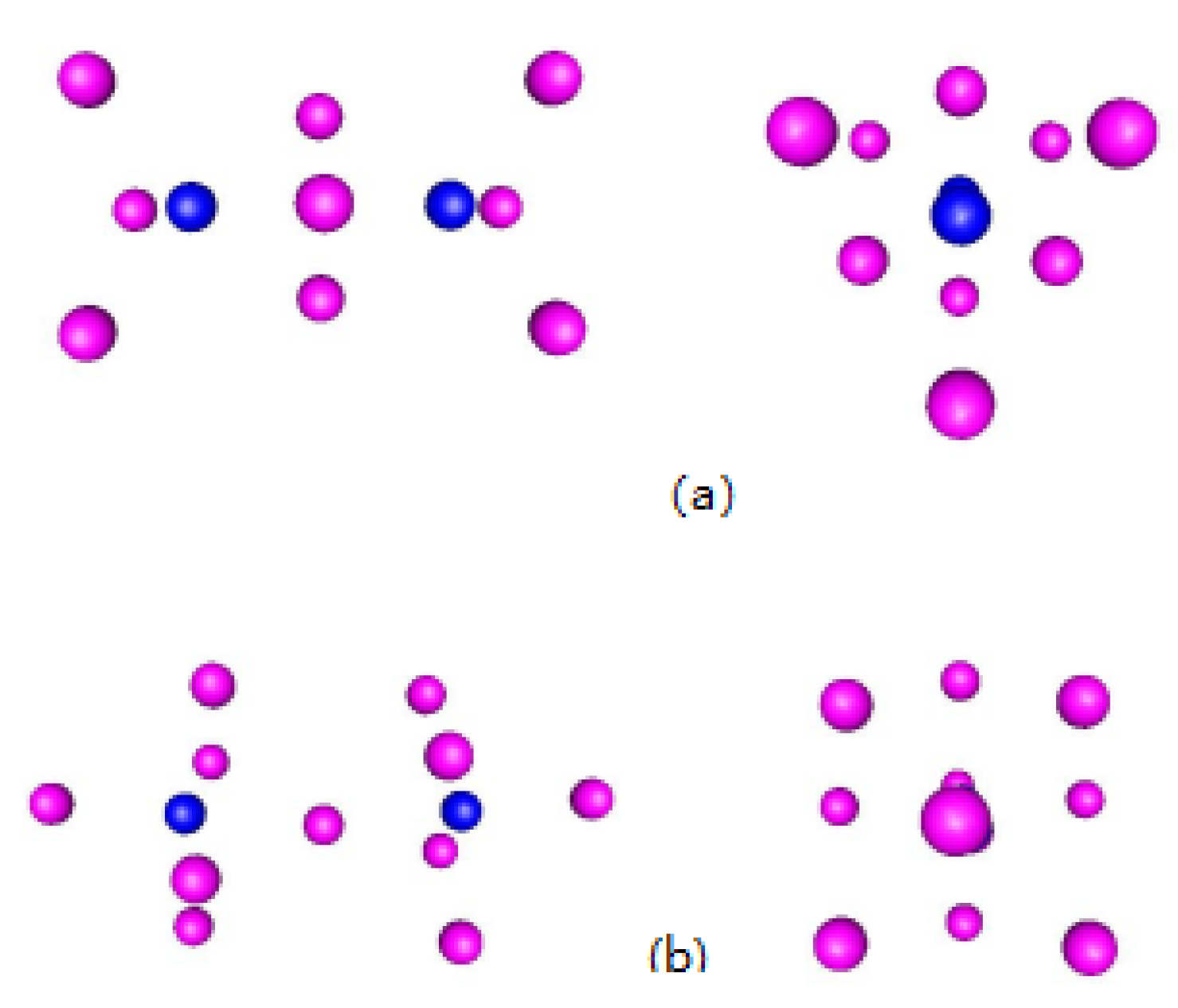
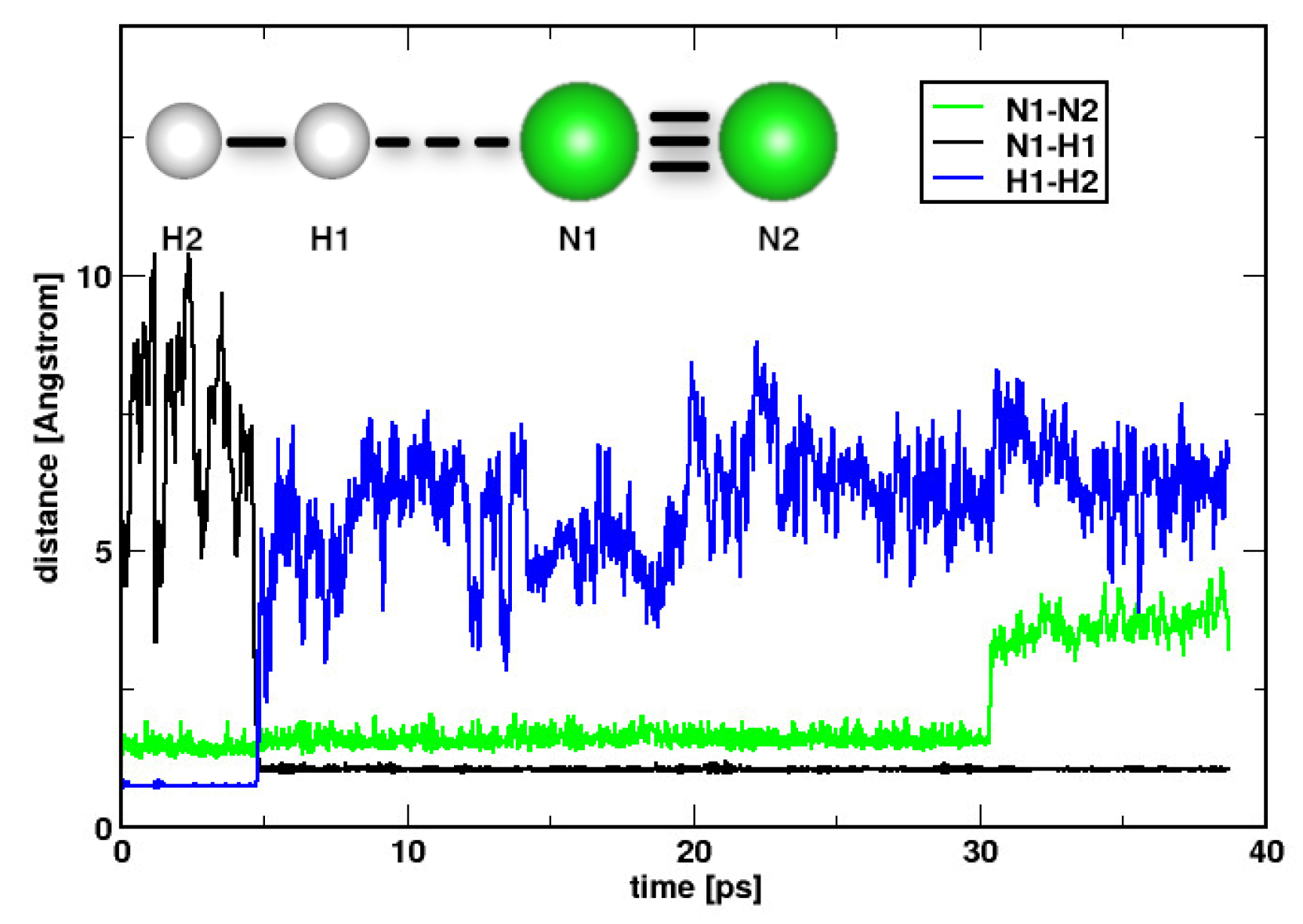
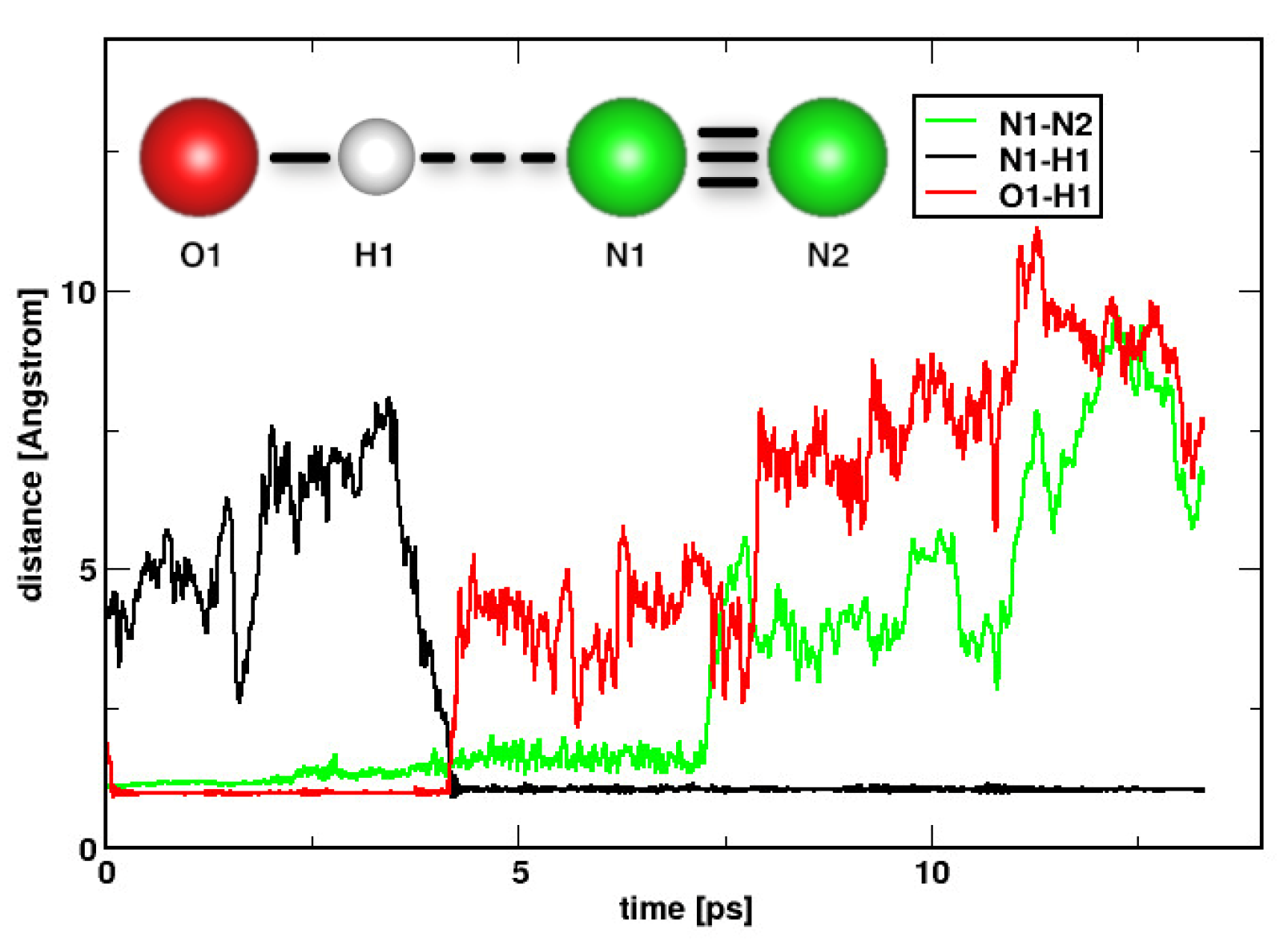
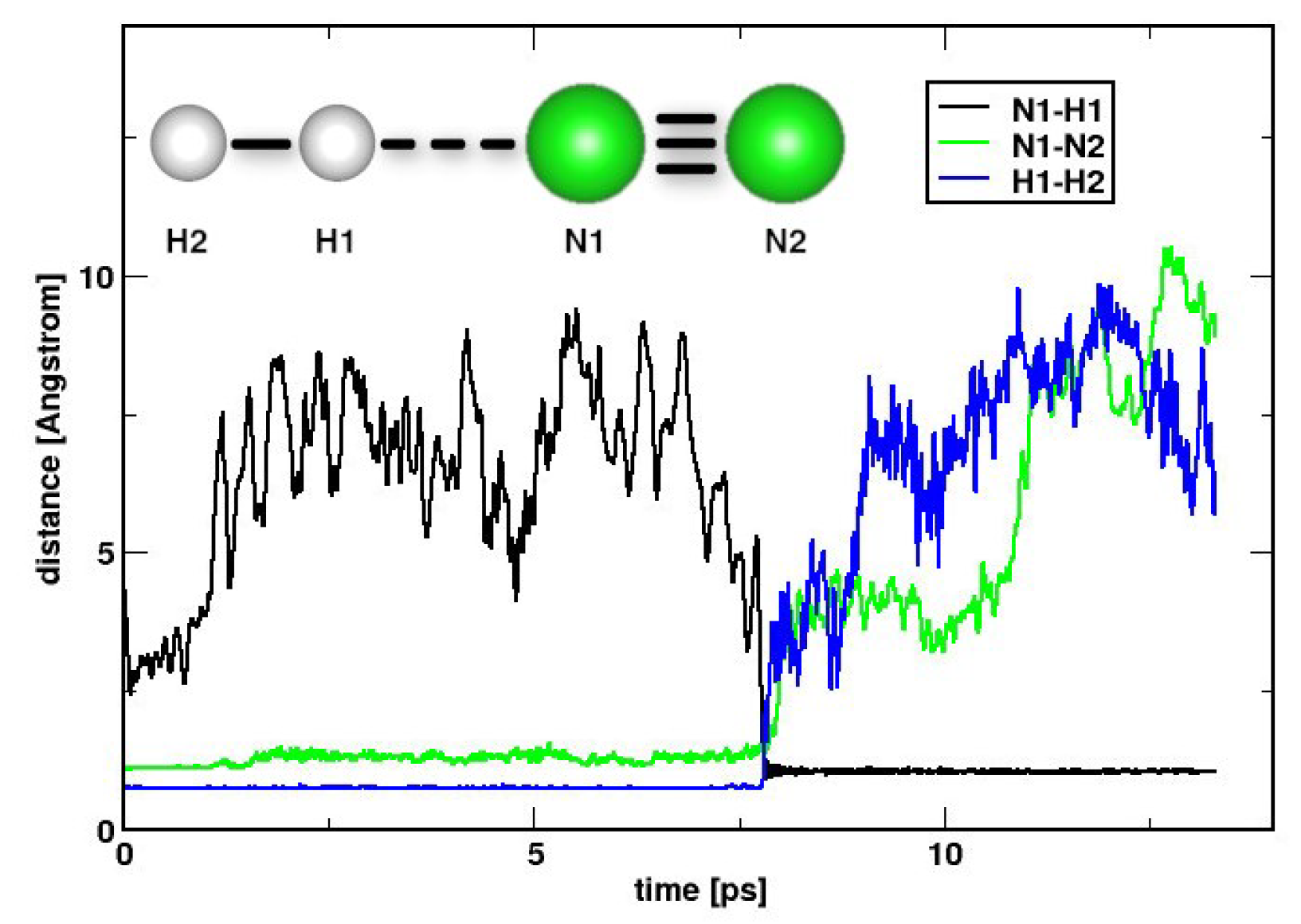

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Simulation | Li/N/H/O | Temp. | N-N | N-H |
|---|---|---|---|---|---|
| Number | [K] | Cleavage | Formation | ||
| Li slab | 1 | 64/0/0/0 | 300 | no | no |
| Li slab | 2 | 64/24/72/0 | 300 | no | no |
| Li slab | 3 | 64/20/68/0 | 300 | yes | no |
| Li slab | 4 | 64/20/64/0 | 300 | yes | no |
| Li slab | 5 | 64/24/72/0 | 600 | yes | no |
| Li slab | 6 | 64/20/68/0 | 600 | yes | no |
| Li slab | 7 | 64/20/64/0 | 600 | yes | no |
| Li slab | 8 | 64/24/72/0 | 800 | yes | no |
| Li slab | 9 | 64/20/68/0 | 800 | yes | no |
| Li slab | 10 | 64/20/64/0 | 800 | yes | yes |
| Li slab | 11 | 64/24/72/0 | 1000 | yes | yes |
| Li slab | 12 | 64/20/68/0 | 1000 | yes | no |
| Li slab | 13 | 64/20/64/0 | 1000 | yes | no |
| Li slab, O | 14 | 64/20/68/12 | 300 | no | no |
| Li slab, O | 15 | 64/12/36/8 | 300 | no | no |
| Li slab, O | 16 | 64/6/18/4 | 300 | no | no |
| Li slab, O | 17 | 64/20/68/12 | 600 | no | no |
| Li slab, O | 18 | 64/12/36/8 | 600 | yes | no |
| Li slab, O | 19 | 64/ 6/18/4 | 600 | yes | no |
| Li slab, O | 20 | 64/20/68/12 | 800 | yes | yes |
| Li slab, O | 21 | 64/12/36/8 | 800 | yes | no |
| Li slab, O | 22 | 64/ 6/18/4 | 800 | no | no |
| Li slab, O | 23 | 64/20/68/12 | 1000 | yes | yes |
| Li slab, O | 24 | 64/12/36/8 | 1000 | yes | yes |
| Li slab, O | 25 | 64/6/18/4 | 1000 | yes | no |
| Method | N-N | Outer Li-N | Inner Li-N |
|---|---|---|---|
| [Å] | [Å] | [Å] | |
| BLYP | 3.04 | 1.85 | 2.02 |
| BLYP/D3BJ | 3.02 | 1.84 | 2.00 |
| B3LYP | 3.03 | 1.84 | 2.00 |
| B3LYP/D3BJ | 3.01 | 1.83 | 1.99 |
| B2PLYP | 3.04 | 1.85 | 2.01 |
| B2PLYP/D3BJ | 3.03 | 1.85 | 2.01 |
| MP2 | 3.11 | 1.87 | 2.06 |
| CCSD | 3.06 | 1.86 | 2.02 |
| Method | N-N | Outer Li-N | Central Li-N | Inner Li-N |
|---|---|---|---|---|
| [Å] | [Å] | [Å] | [Å] | |
| BLYP | 3.96 | 1.85 | 1.91–1.94 | 2.00 |
| BLYP/D3BJ | 3.89 | 1.84 | 1.91–1.93 | 1.95 |
| B3LYP | 3.95 | 1.84 | 1.90–1.92 | 1.98 |
| B3LYP/D3BJ | 3.89 | 1.83 | 1.90–1.91 | 1.94 |
| B2PLYP | 3.98 | 1.84 | 1.92–1.93 | 1.99 |
| B2PLYP/D3BJ | 3.94 | 1.84 | 1.92–1.93 | 1.97 |
| MP2 | 4.04 | 1.87 | 1.94–1.95 | 2.02 |
| CCSD | 4.01 | 1.85 | 1.93–1.94 | 2.01 |
| Simulation | Atom | Coordination |
|---|---|---|
| 10 | N | 6 |
| 10 | N | 2 |
| 10 | N | 6 |
| 10 | N | 3 |
| 11 | N | 6 |
| 11 | N | 3 |
| 20 | O | 3 |
| 20 | N | 3 |
| 20 | N | 5 |
| 23 | N | 3 |
| 23 | N | 4 |
| 23 | O | 4 |
| 23 | N | 3 |
| 23 | N | 3 |
| 24 | N | 4 |
| 24 | N | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniscalco, D.; Rudolph, D.A.; Nadimi, E.; Frank, I. The First Reaction Steps of Lithium-Mediated Ammonia Synthesis: Ab Initio Simulation. Nitrogen 2022, 3, 404-413. https://doi.org/10.3390/nitrogen3030026
Maniscalco D, Rudolph DA, Nadimi E, Frank I. The First Reaction Steps of Lithium-Mediated Ammonia Synthesis: Ab Initio Simulation. Nitrogen. 2022; 3(3):404-413. https://doi.org/10.3390/nitrogen3030026
Chicago/Turabian StyleManiscalco, Dominykas, Dominik A. Rudolph, Ebrahim Nadimi, and Irmgard Frank. 2022. "The First Reaction Steps of Lithium-Mediated Ammonia Synthesis: Ab Initio Simulation" Nitrogen 3, no. 3: 404-413. https://doi.org/10.3390/nitrogen3030026
APA StyleManiscalco, D., Rudolph, D. A., Nadimi, E., & Frank, I. (2022). The First Reaction Steps of Lithium-Mediated Ammonia Synthesis: Ab Initio Simulation. Nitrogen, 3(3), 404-413. https://doi.org/10.3390/nitrogen3030026