TranScreen: Transfer Learning on Graph-Based Anti-Cancer Virtual Screening Model

 ,
,
Abstract
1. Introduction
- TranScreen pipeline: A practical pipeline is developed, which enables the usage of graph convolutional neural networks (GCNNs) for virtual screening and transferring the learned knowledge between multiple molecular datasets.
- Creation of a collection of weights, which can be used as network initializations.
- Comparing three methods of ranking models before fine-tuning takes place to select the model for future tasks.
2. Materials and Methods
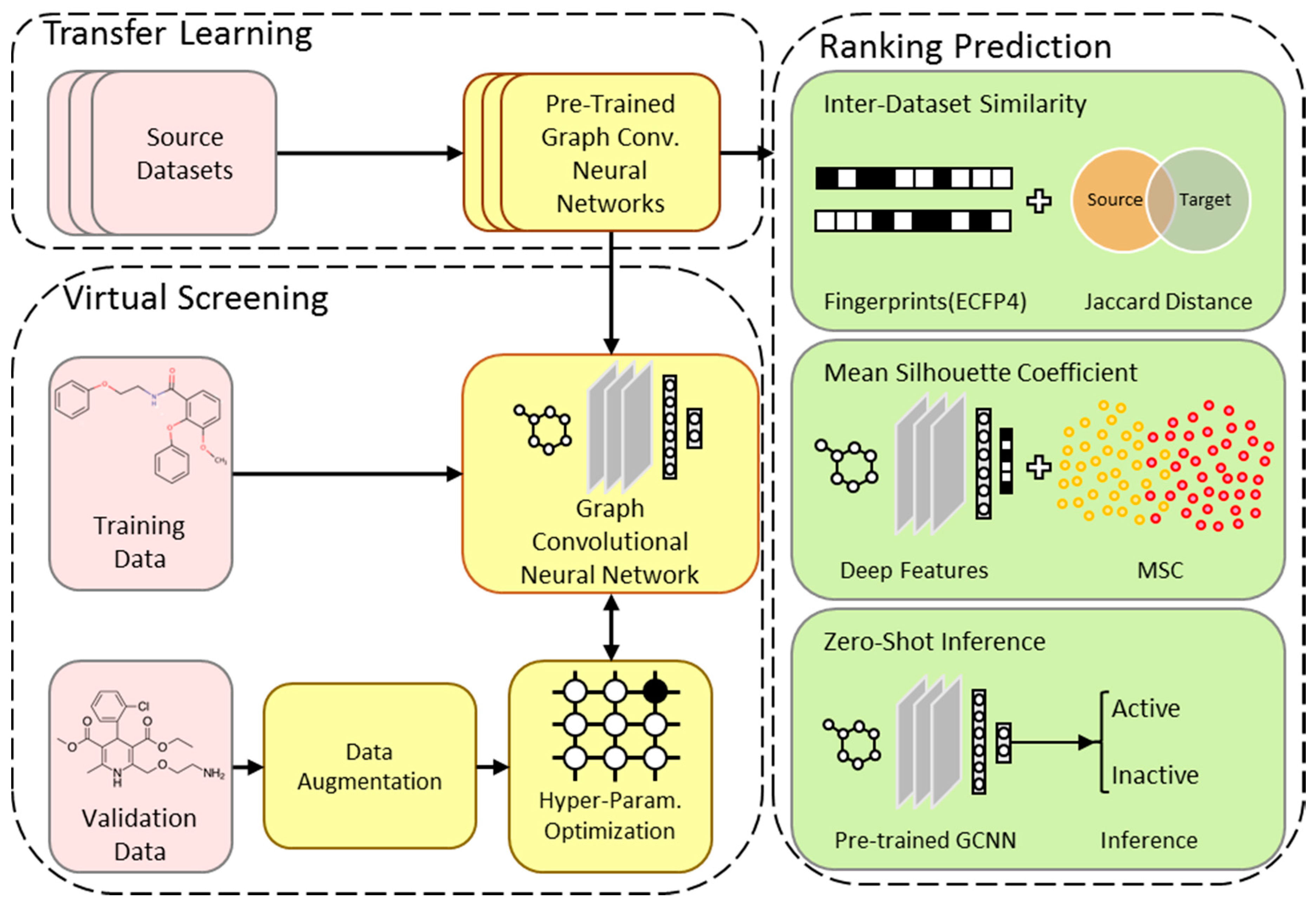
2.1. Overview of the TranScreen Pipeline
2.2. Data
2.2.1. Source Data
2.2.2. Target Data
2.2.3. Data Preprocessing and Partitioning
2.3. Model Creation and Training
2.3.1. Graph Convolutional Neural Networks
2.3.2. Common Network Architecture
2.3.3. Baseline Model and Internal Validation
2.4. Transfer Learning and Fine-Tuning
2.5. Model Rank Prediction Before Fine-Tuning
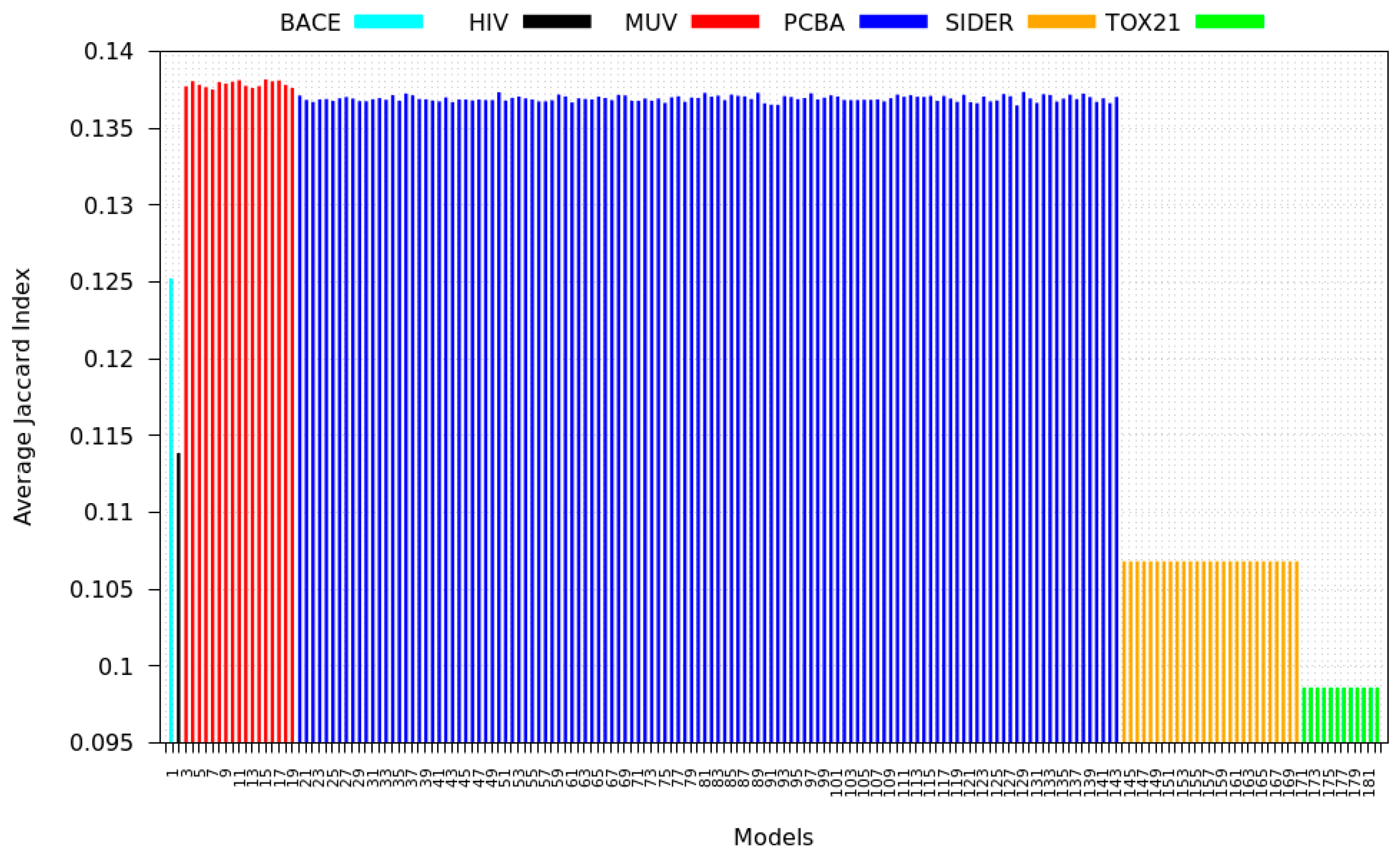
2.5.1. Inter-Dataset Similarity Comparison
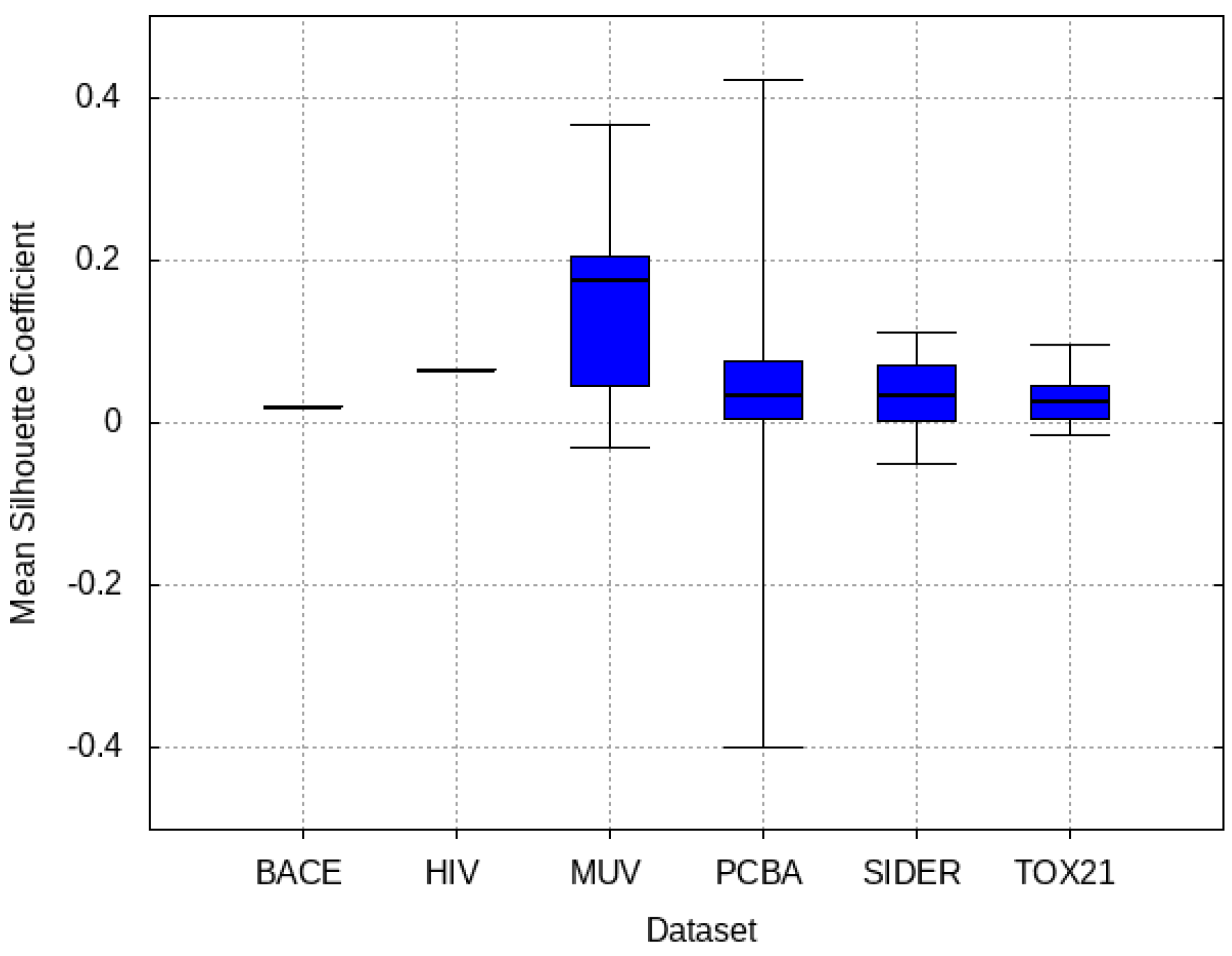
2.5.2. Mean Silhouette Coefficient on Deep Features
2.5.3. Zero-Shot Inference
2.6. Evaluation
3. Results
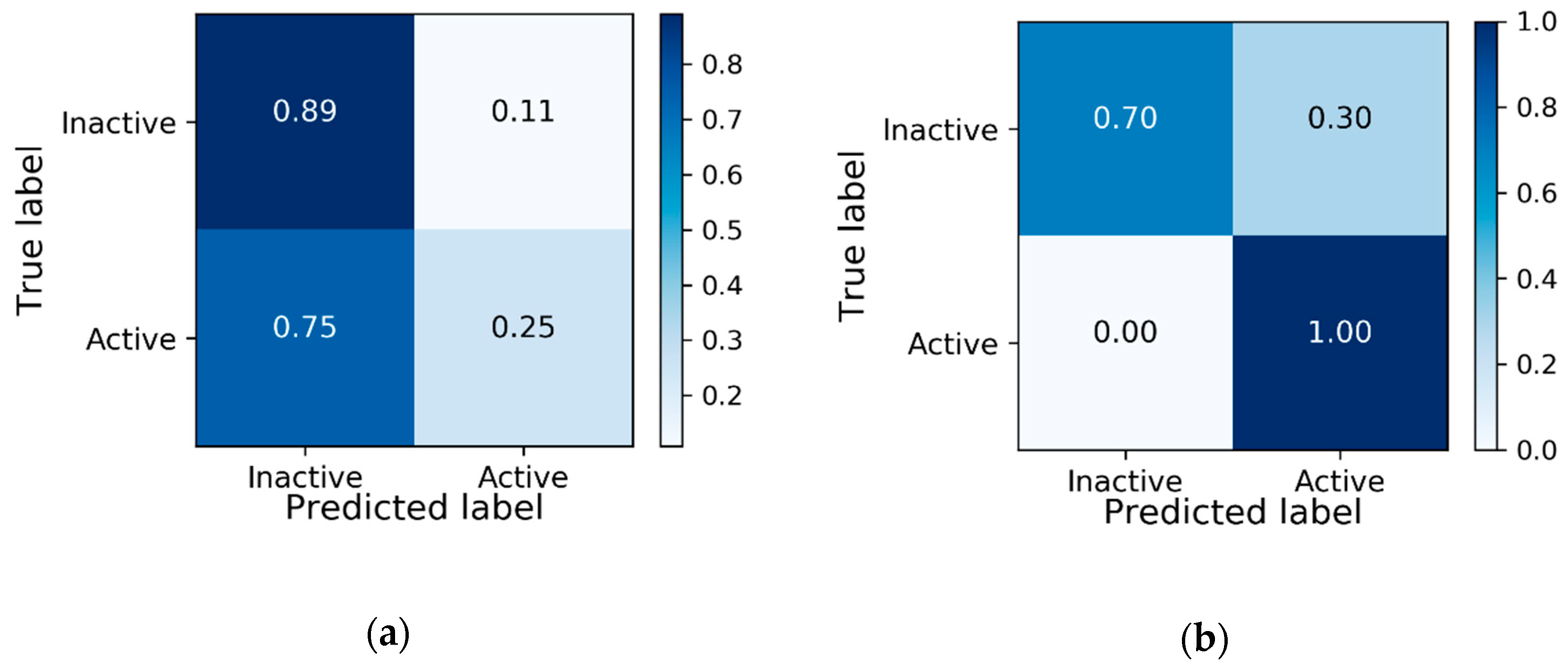
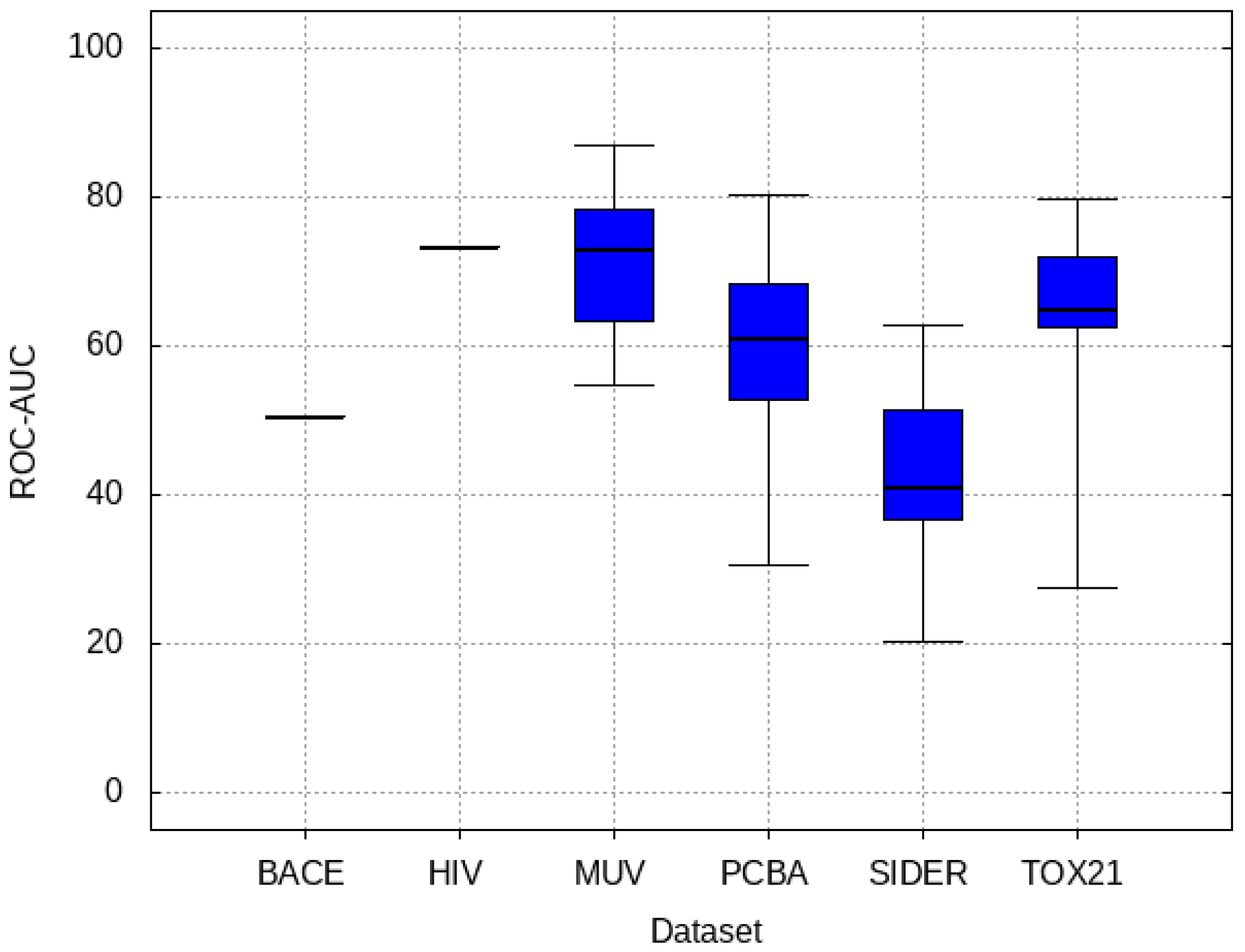
3.1. Baseline Model Results
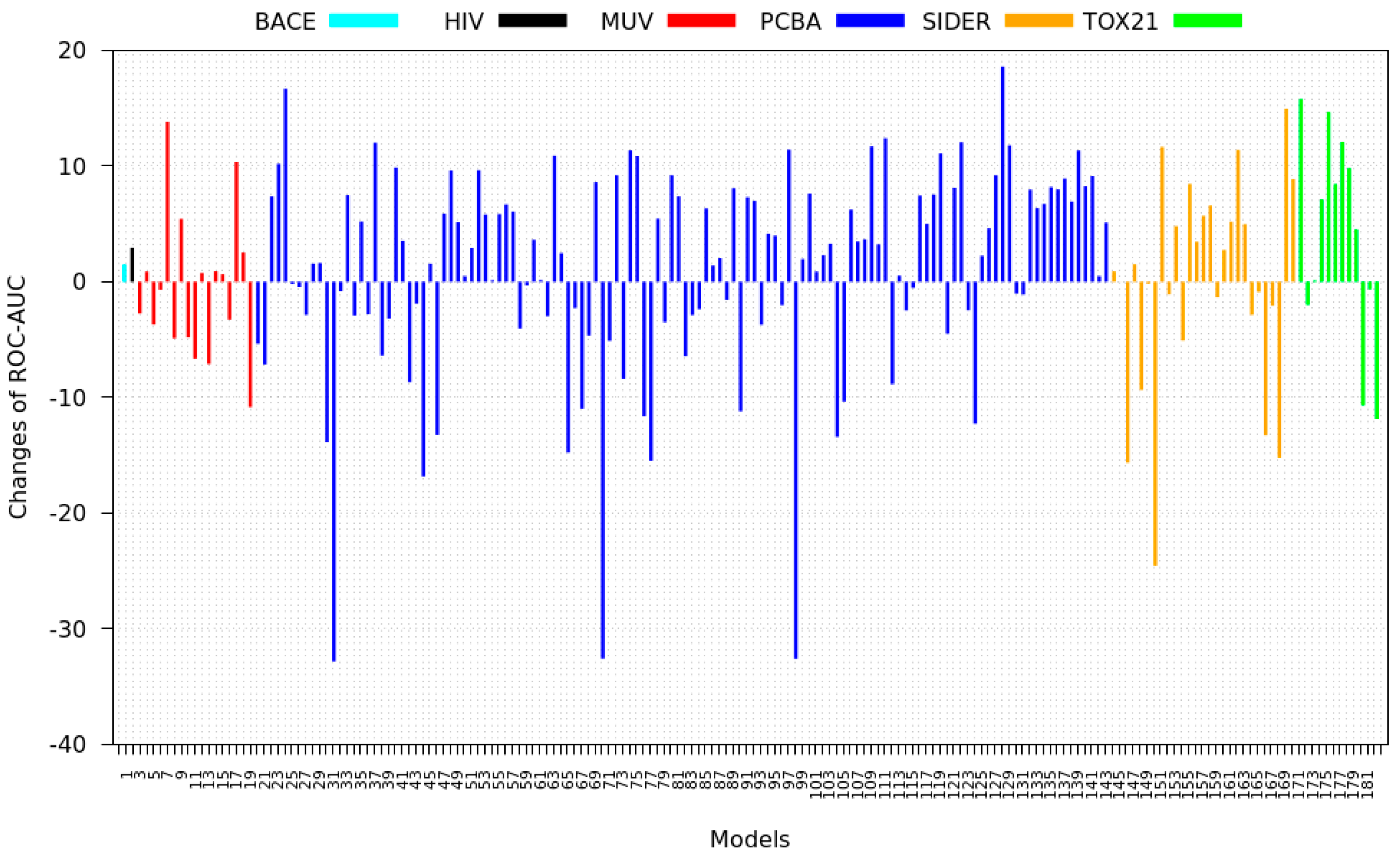
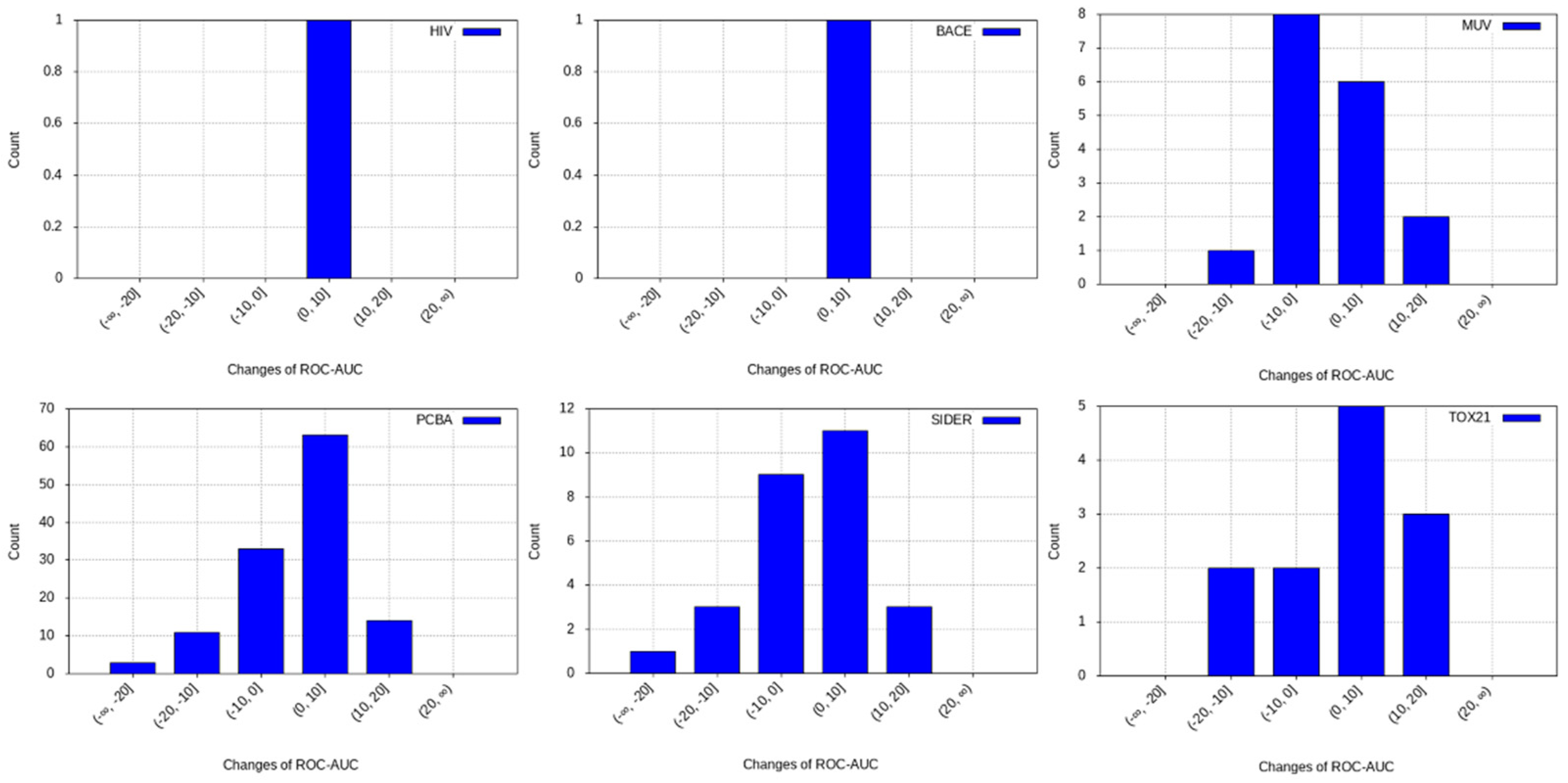
3.2. Transfer Learning Results
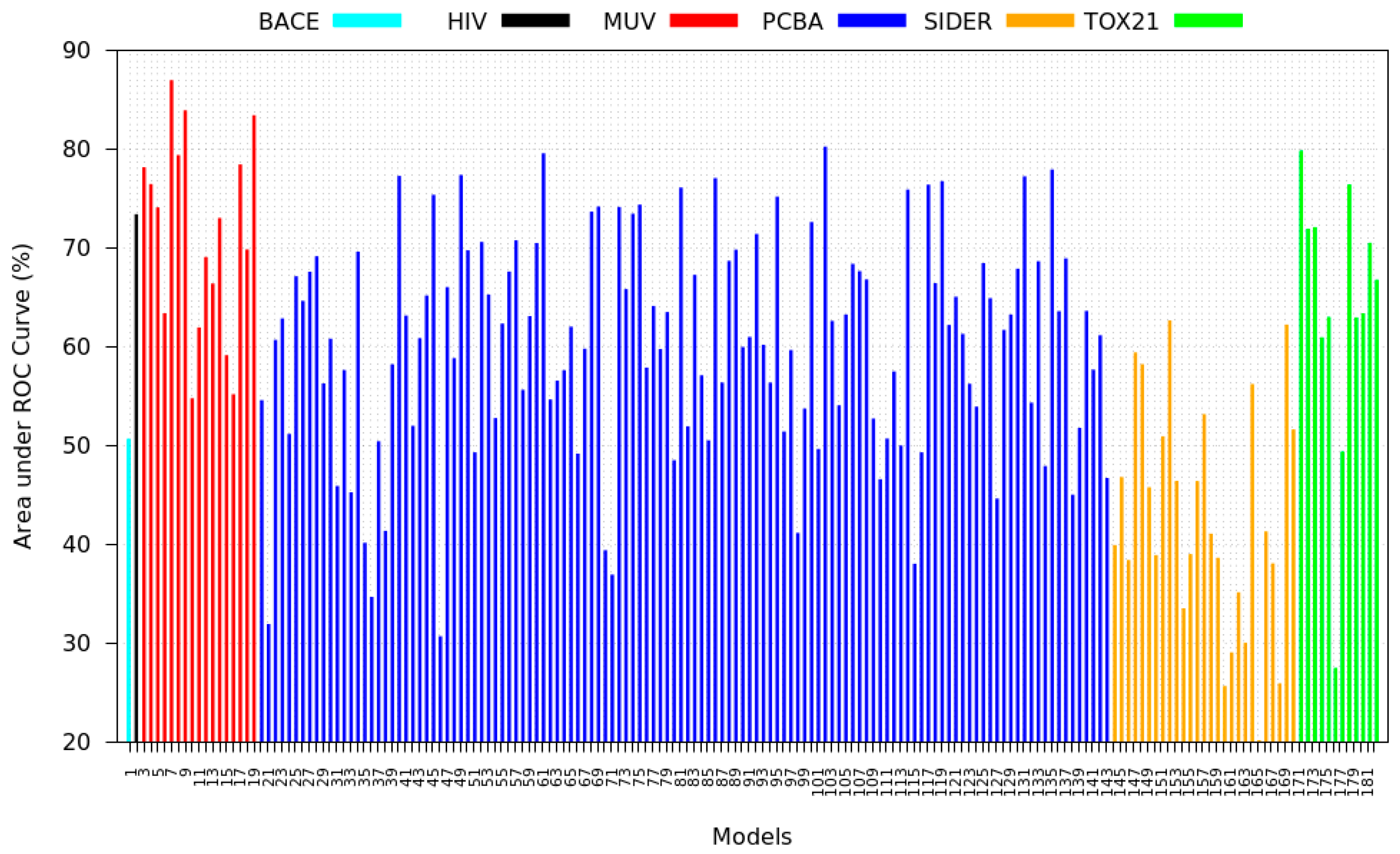
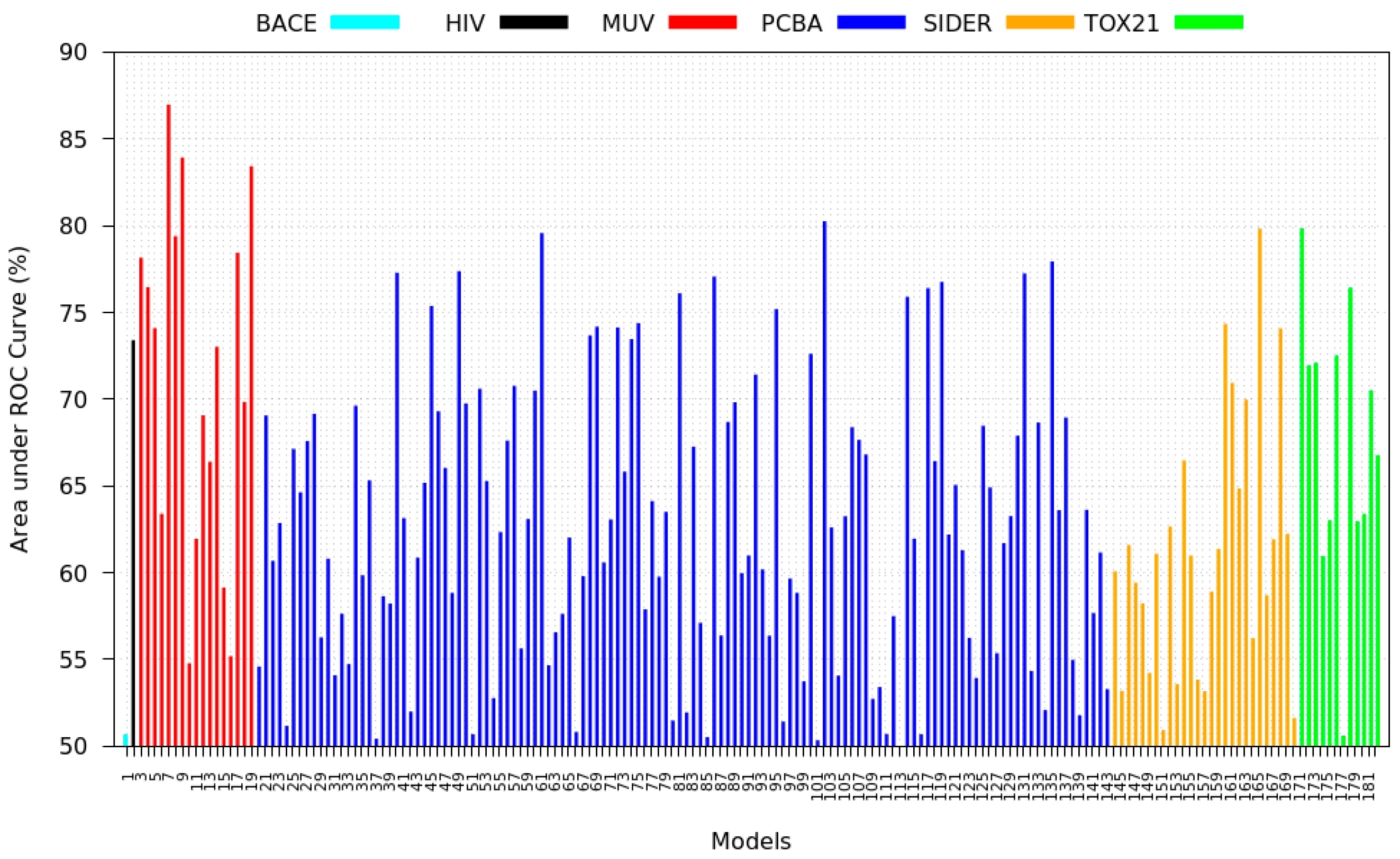
3.3. Inter-Dataset Similarity Results
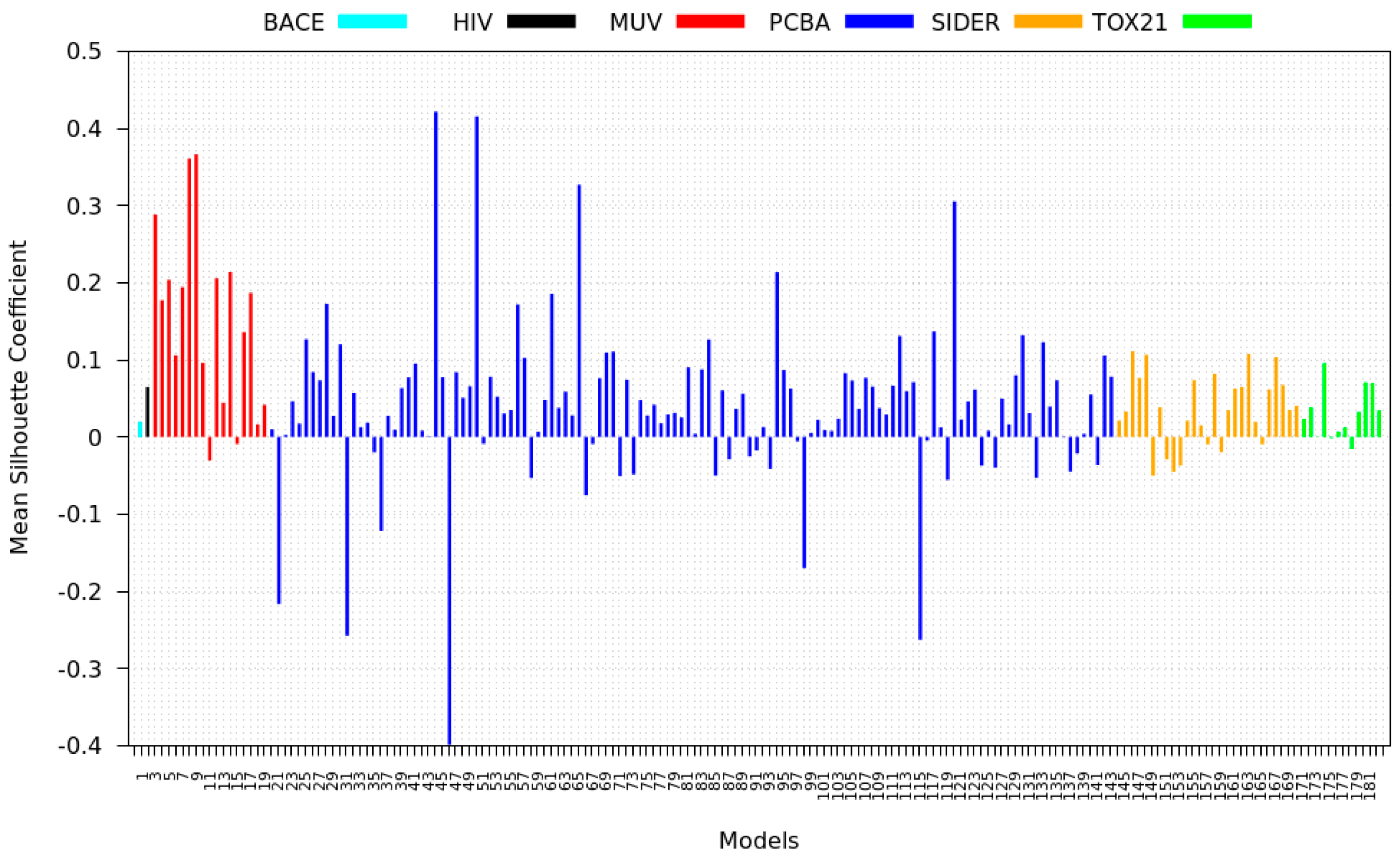
3.4. Mean Silhouette Coefficient Results
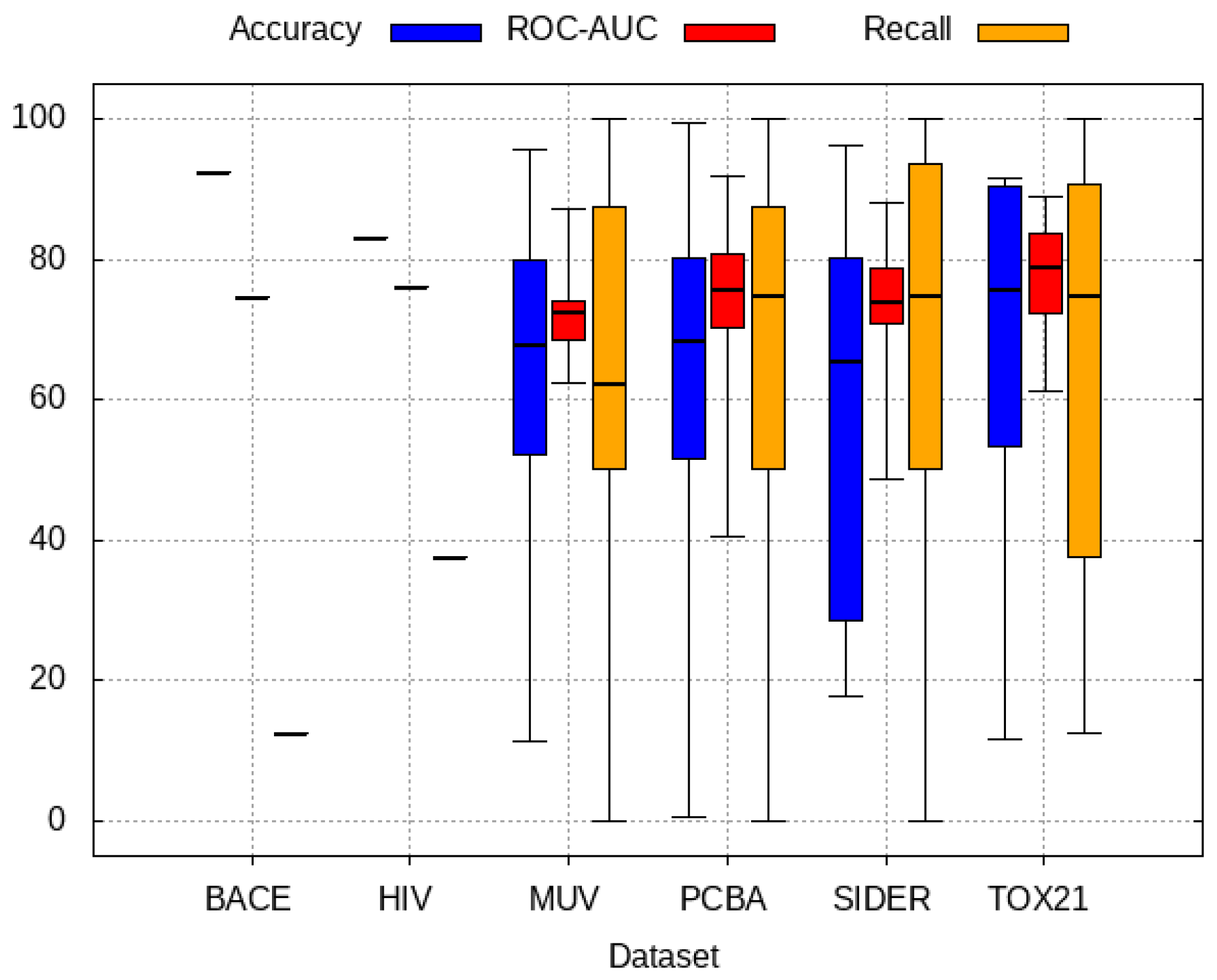
3.5. Zero-Shot Inference Results
3.6. Model Rank Prediction Results
4. Discussion and Results Interpretations
- PCBA: This dataset is one of the closest and most similar (in terms of fingerprint similarity) to the target dataset. It has the highest MSC, indicating that deep features learned from this data source can distinguish between active and inactive molecules. The best performing data source belongs to this dataset. However, it also possesses the tasks that yielded the lowest MSC and the worst performance.
- MUV: This dataset is also very similar to the target dataset. On average the models trained on this dataset delivered the highest MSC. However, on average these models yielded the lowest performance improvement.
- Tox21: This dataset is the most dissimilar to the target dataset. It does not perform well when tested with MSC measurement. However, the models from this dataset deliver the highest average improvement after transfer learning.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Cancer and p53
Appendix B








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Values | Parameter | Value |
|---|---|---|---|
| Number of conv. layers | 3 | Size of conv. layers | 64 |
| Number of neurons | 256 | Learning rate | 0.0001 |
| Dropout | 0 | Batch size | 128 |
| Task | Number | Task | Number | Task | Number | Task | Number |
|---|---|---|---|---|---|---|---|
| BACE | 1 | PCBA-885 | 36 | PCBA-485281 | 71 | PCBA-588590 | 106 |
| HIV | 2 | PCBA-887 | 37 | PCBA-485290 | 72 | PCBA-588591 | 107 |
| MUV-466 | 3 | PCBA-891 | 38 | PCBA-485294 | 73 | PCBA-588795 | 108 |
| MUV-733 | 4 | PCBA-899 | 39 | PCBA-485297 | 74 | PCBA-588855 | 109 |
| MUV-737 | 5 | PCBA-912 | 40 | PCBA-485313 | 75 | PCBA-602179 | 110 |
| MUV-810 | 6 | PCBA-914 | 41 | PCBA-485314 | 76 | PCBA-1460 | 111 |
| MUV-832 | 7 | PCBA-915 | 42 | PCBA-485341 | 77 | PCBA-602233 | 112 |
| MUV-846 | 8 | PCBA-1479 | 43 | PCBA-1454 | 78 | PCBA-602310 | 113 |
| MUV-852 | 9 | PCBA-925 | 44 | PCBA-485349 | 79 | PCBA-602313 | 114 |
| MUV-858 | 10 | PCBA-926 | 45 | PCBA-485353 | 80 | PCBA-602332 | 115 |
| MUV-859 | 11 | PCBA-927 | 46 | PCBA-485360 | 81 | PCBA-624170 | 116 |
| MUV-548 | 12 | PCBA-938 | 47 | PCBA-485364 | 82 | PCBA-624171 | 117 |
| MUV-600 | 13 | PCBA-995 | 48 | PCBA-485367 | 83 | PCBA-624173 | 118 |
| MUV-644 | 14 | PCBA-1631 | 49 | PCBA-492947 | 84 | PCBA-624202 | 119 |
| MUV-652 | 15 | PCBA-1634 | 50 | PCBA-493208 | 85 | PCBA-624246 | 120 |
| MUV-689 | 16 | PCBA-1688 | 51 | PCBA-504327 | 86 | PCBA-624287 | 121 |
| MUV-692 | 17 | PCBA-1721 | 52 | PCBA-504332 | 87 | PCBA-1461 | 122 |
| MUV-712 | 18 | PCBA-2100 | 53 | PCBA-504333 | 88 | PCBA-624288 | 123 |
| MUV-713 | 19 | PCBA-2101 | 54 | PCBA-1457 | 89 | PCBA-624291 | 124 |
| PCBA-1030 | 20 | PCBA-2147 | 55 | PCBA-504339 | 90 | PCBA-624296 | 125 |
| PCBA-1469 | 21 | PCBA-1379 | 56 | PCBA-504444 | 91 | PCBA-624297 | 126 |
| PCBA-720553 | 22 | PCBA-2242 | 57 | PCBA-504466 | 92 | PCBA-624417 | 127 |
| PCBA-720579 | 23 | PCBA-2326 | 58 | PCBA-504467 | 93 | PCBA-651635 | 128 |
| PCBA-720580 | 24 | PCBA-2451 | 59 | PCBA-504706 | 94 | PCBA-651644 | 129 |
| PCBA-720707 | 25 | PCBA-2517 | 60 | PCBA-504842 | 95 | PCBA-651768 | 130 |
| PCBA-720708 | 26 | PCBA-2528 | 61 | PCBA-504845 | 96 | PCBA-651965 | 131 |
| PCBA-720709 | 27 | PCBA-2546 | 62 | PCBA-504847 | 97 | PCBA-652025 | 132 |
| PCBA-720711 | 28 | PCBA-2549 | 63 | PCBA-504891 | 98 | PCBA-1468 | 133 |
| PCBA-743255 | 29 | PCBA-2551 | 64 | PCBA-540276 | 99 | PCBA-652104 | 134 |
| PCBA-743266 | 30 | PCBA-2662 | 65 | PCBA-1458 | 100 | PCBA-652105 | 135 |
| PCBA-875 | 31 | PCBA-2675 | 66 | PCBA-540317 | 101 | PCBA-652106 | 136 |
| PCBA-1471 | 32 | PCBA-1452 | 67 | PCBA-588342 | 102 | PCBA-686970 | 137 |
| PCBA-881 | 33 | PCBA-2676 | 68 | PCBA-588453 | 103 | PCBA-686978 | 138 |
| PCBA-883 | 34 | PCBA-411 | 69 | PCBA-588456 | 104 | PCBA-686979 | 139 |
| PCBA-884 | 35 | PCBA-463254 | 70 | PCBA-588579 | 105 | PCBA-720504 | 140 |
| Task | Number | Task | Number | Task | Number | Task | Number |
|---|---|---|---|---|---|---|---|
| PCBA-720532 | 141 | Metabolism and nutrition disorders | 152 | Vascular disorders | 163 | SR-p53 | 174 |
| PCBA-720542 | 142 | Musculoskeletal and connective tissue disorders | 153 | “Neoplasms benign, malignant and unspecified (incl cysts and polyps)” | 164 | NR-AR | 175 |
| PCBA-720551 | 143 | Nervous system disorders | 154 | “Pregnancy, puerperium and perinatal conditions” | 165 | NR-AR-LBD | 176 |
| “Congenital, familial and genetic disorders” | 144 | “Injury, poisoning and procedural complications” | 155 | “Respiratory, thoracic and mediastinal disorders” | 166 | NR-Aromatase | 177 |
| Eye disorders | 145 | Product issues | 156 | Blood and lymphatic system disorders | 167 | NR-ER | 178 |
| Gastrointestinal disorders | 146 | Psychiatric disorders | 157 | Cardiac disorders | 168 | NR-ER-LBD | 179 |
| General disorders and administration site conditions | 147 | Renal and urinary disorders | 158 | Ear and labyrinth disorders | 169 | NR-PPAR-gamma | 180 |
| Hepatobiliary disorders | 148 | Reproductive system and breast disorders | 159 | Endocrine disorders | 170 | SR-ARE | 181 |
| Immune system disorders | 149 | Skin and subcutaneous tissue disorders | 160 | NR-AhR | 171 | SR-ATAD5 | 182 |
| Infections and infestations | 150 | Social circumstances | 161 | SR-HSE | 172 | ||
| Investigations | 151 | Surgical and medical procedures | 162 | SR-MMP | 173 |
References
- Carnero, A. High throughput screening in drug discovery. Clin. Transl. Oncol. 2006, 8, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. (N. Y.) 2017, 3, 651–657. (In English) [Google Scholar] [CrossRef] [PubMed]
- Miljković, F.; Rodríguez-Pérez, R.; Bajorath, J. Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, A.; Pérez-Castillo, Y.; Schürer, S.C.; Nicolotti, O.; Mangiatordi, G.F.; Borges, F.; Cordeiro, M.N.D.; Tejera, E.; Medina-Franco, J.L.; Cruz-Monteagudo, M. From flamingo dance to (desirable) drug discovery: A nature-inspired approach. Drug Discov. Today 2017, 22, 1489–1502. [Google Scholar]
- Cruz-Monteagudo, M.; Ancede-Gallardo, E.; Jorge, M.; Cordeiro, M.N.D.S. Chemoinformatics Profiling of Ionic Liquids—Automatic and Chemically Interpretable Cytotoxicity Profiling, Virtual Screening, and Cytotoxicophore Identification. Toxicol. Sci. 2013, 136, 548–565. [Google Scholar] [CrossRef][Green Version]
- Perez-Castillo, Y.; Sánchez-Rodríguez, A.; Tejera, E.; Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.; Le-Thi-Thu, H.; Pham-The, H. A desirability-based multi objective approach for the virtual screening discovery of broad-spectrum anti-gastric cancer agents. PLoS ONE 2018, 13, e0192176. [Google Scholar] [CrossRef]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Minnich, A.J.; McLoughlin, K.; Tse, M.; Deng, J.; Weber, A.; Murad, N.; Madej, B.D.; Ramsundar, B.; Rush, T.; Calad-Thomson, S.; et al. AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 1955–1968. [Google Scholar] [CrossRef]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sianes, J.; Pérez-Sánchez, H.; Díaz, F. Virtual Screening: A Challenge for Deep Learning. In 10th International Conference on Practical Applications of Computational Biology & Bioinformatics; Springer International Publishing: Cham, Switzerland, 2016; pp. 13–22. [Google Scholar]
- Fischer, B.; Merlitz, H.; Wenzel, W. Increasing Diversity in In-silico Screening with Target Flexibility. In Computational Life Sciences; Springer: Berlin/Heidelberg, Germany, 2005; pp. 186–197. [Google Scholar]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. Comparison of Fingerprint-Based Methods for Virtual Screening Using Multiple Bioactive Reference Structures. J. Chem. Inf. Comput. Sci. 2004, 44, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Ramsundar, B.; Kearnes, S.; Riley, P.; Webster, D.; Konerding, D.; Pande, V. Massively multitask networks for drug discovery. arXiv 2015, arXiv:1502.02072. [Google Scholar]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for Pre-training graph neural networks. arXiv 2019, arXiv:1905.12265. [Google Scholar]
- Liu, S. Exploration on Deep Drug Discovery: Representation and Learning; Computer Science, University of Wisconsin-Madison: Madison, WI, USA, 2018. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- PubChem Database. Source=NCGC AID=904. 2007. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/904 (accessed on 18 May 2020).
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Torng, W.; Altman, R.B. Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model. 2019, 59, 4131–4149. [Google Scholar] [CrossRef]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. [Google Scholar] [CrossRef]
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V.; Leswing, K.; Wu, Z. Deep Learning for the Life Sciences; O’Reilly Media: Sebastopol, CA, USA, 2019. [Google Scholar]
- Bjerrum, E.J. Smiles enumeration as data augmentation for neural network modeling of molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Arshadi, A.K.; Salem, M.; Collins, J.; Yuan, J.S.; Chakrabarti, D. DeepMalaria: Artificial Intelligence Driven Discovery of Potent Antiplasmodials. Front. Pharmacol. 2019, 10, 1526. [Google Scholar] [CrossRef] [PubMed]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Nassif, A.B.; Shahin, I.; Attili, I.; Azzeh, M.; Shaalan, K. Speech Recognition Using Deep Neural Networks: A Systematic Review. IEEE Access 2019, 7, 19143–19165. [Google Scholar] [CrossRef]
- Boumi, S.; Vela, A.; Chini, J. Quantifying the relationship between student enrollment patterns and student performance. arXiv 2020, arXiv:2003.10874. [Google Scholar]
- Zhang, K.; Guo, Y.; Wang, X.; Yuan, J.; Ding, Q. Multiple Feature Reweight DenseNet for Image Classification. IEEE Access 2019, 7, 9872–9880. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, Y.; Chua, T.-S.; Schiele, B. Meta-transfer learning for few-shot learning. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 403–412. [Google Scholar]
- Liu, S.; Johns, E.; Davison, A.J. End-to-end multi-task learning with attention. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 1871–1880. [Google Scholar]
- Zhuang, F.; Qi, Z.; Duan, K.; Xi, D.; Zhu, Y.; Zhu, H.; Xiong, H.; He, Q. A Comprehensive Survey on Transfer Learning. arXiv 2019, arXiv:1911.02685. [Google Scholar]
- Frankle, J.; Carbin, M. The lottery ticket hypothesis: Finding sparse trainable neural networks. arXiv 2018, arXiv:1803.03635. [Google Scholar]
- Fawaz, H.I.; Forestier, G.; Weber, J.; Idoumghar, L.; Muller, P. Transfer learning for time series classification. In Proceedings of the 2018 IEEE International Conference on Big Data (Big Data), Zurich, Switzerland Seattle, WA, USA, 10–13 December 2018; pp. 1367–1376. [Google Scholar]
- Wang, M.; Deng, W. Deep visual domain adaptation: A survey. Neurocomputing 2018, 312, 135–153. [Google Scholar] [CrossRef]
- Zhang, H.; Koniusz, P. Model Selection for Generalized Zero-Shot Learning. In Computer Vision—ECCV 2018 Workshops; Springer International Publishing: Cham, Switzerland, 2019; pp. 198–204. [Google Scholar]
- Zhang, H.; Koniusz, P. Zero-Shot Kernel Learning. In Proceedings of the 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–22 June 2018; pp. 7670–7679. [Google Scholar]
- Ben-David, S.; Blitzer, J.; Crammer, K.; Pereira, F. Analysis of representations for domain adaptation. In Advances in NEURAL Information Processing Systems; The MIT Press: Cambridge, MA, USA, 2007; pp. 137–144. [Google Scholar]
- Meiseles, A.; Rokach, L. Source Model Selection for Deep Learning in the Time Series Domain. IEEE Access 2020, 8, 6190–6200. [Google Scholar] [CrossRef]
- Liu, S.; Alnammi, M.; Ericksen, S.S.; Voter, A.F.; Ananiev, G.E.; Keck, J.L.; Hoffmann, F.M.; Wildman, S.A.; Gitter, A. Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model. 2019, 59, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Swamidass, S.J.; Azencott, C.-A.; Lin, T.-W.; Gramajo, H.; Tsai, S.-C.; Baldi, P. Influence relevance voting: An accurate and interpretable virtual high throughput screening method. (in eng). J. Chem. Inf. Model. 2009, 49, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Koniusz, P. Power Normalizing Second-Order Similarity Network for Few-Shot Learning. In Proceedings of the 2019 IEEE Winter Conference on Applications of Computer Vision (WACV), Waikoloa Village, HI, USA, 7–11 January 2019; pp. 1185–1193. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Yabroff, K.R.; Warren, J.L.; Brown, M.L. Costs of cancer care in the USA: A descriptive review. Nat. Clin. Pract. Oncol. 2007, 4, 643–656. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef]
- Wang, L.H.; Wu, C.F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Ashcroft, M.; Taya, Y.; Vousden, K.H. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell Biol. 2000, 20, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef]








| Dataset | Data Type | Number of Tasks | Number of Compounds |
|---|---|---|---|
| PCBA | SMILES | 124 | 437,929 |
| MUV | 17 | 93,087 | |
| HIV | 1 | 41,127 | |
| BACE | 1 | 1513 | |
| Tox21 | 12 | 7831 | |
| SIDER | 27 | 1427 |
| Dataset | Data Type | Number of Tasks | Number of Compounds | Number of Active Compounds |
|---|---|---|---|---|
| PCBA-904 | SMILES | 1 | 437,929 | 528 |
| Task | Partition | Accuracy | Recall | ROC-AUC |
|---|---|---|---|---|
| PCBA-904 | Validation | 90.96 | 57.14 | 0.7846 |
| PCBA-904 | Test | 89.66 | 25 | 0.7322 |
| Source Dataset | Source Task | Target Task | Accuracy | Recall | BEDROC (Alpha = 1) | ROC-AUC | Improvement |
|---|---|---|---|---|---|---|---|
| PCBA | PCBA-651635 | PCBA-904 | 71.48 | 100 | 0.779 | 0.918 | 0.186 |
| Tox21 | NR-AhR | 82.11 | 75 | 0.678 | 0.889 | 0.158 | |
| SIDER | Ear and labyrinth disorders | 73.56 | 87.5 | 0.392 | 0.882 | 0.150 | |
| MUV | MUV-832 | 81.68 | 75 | 0.677 | 0.871 | 0.138 | |
| HIV | HIV | 83.05 | 37.5 | 0.505 | 0.761 | 0.029 | |
| BACE | BACE | 92.31 | 12.5 | 0.399 | 0.747 | 0.015 | |
| PCBA [18] | PCBA-903 | - | - | - | 0.81 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, M.; Khormali, A.; Arshadi, A.K.; Webb, J.; Yuan, J.-S. TranScreen: Transfer Learning on Graph-Based Anti-Cancer Virtual Screening Model. Big Data Cogn. Comput. 2020, 4, 16. https://doi.org/10.3390/bdcc4030016
Salem M, Khormali A, Arshadi AK, Webb J, Yuan J-S. TranScreen: Transfer Learning on Graph-Based Anti-Cancer Virtual Screening Model. Big Data and Cognitive Computing. 2020; 4(3):16. https://doi.org/10.3390/bdcc4030016
Chicago/Turabian StyleSalem, Milad, Aminollah Khormali, Arash Keshavarzi Arshadi, Julia Webb, and Jiann-Shiun Yuan. 2020. "TranScreen: Transfer Learning on Graph-Based Anti-Cancer Virtual Screening Model" Big Data and Cognitive Computing 4, no. 3: 16. https://doi.org/10.3390/bdcc4030016
APA StyleSalem, M., Khormali, A., Arshadi, A. K., Webb, J., & Yuan, J.-S. (2020). TranScreen: Transfer Learning on Graph-Based Anti-Cancer Virtual Screening Model. Big Data and Cognitive Computing, 4(3), 16. https://doi.org/10.3390/bdcc4030016