



In Silico Analysis Reveals High Levels of Genetic Diversity of Plasmodium knowlesi Cell Traversal Protein for Ookinetes and Sporozoites (PkCelTOS) in Clinical Samples

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
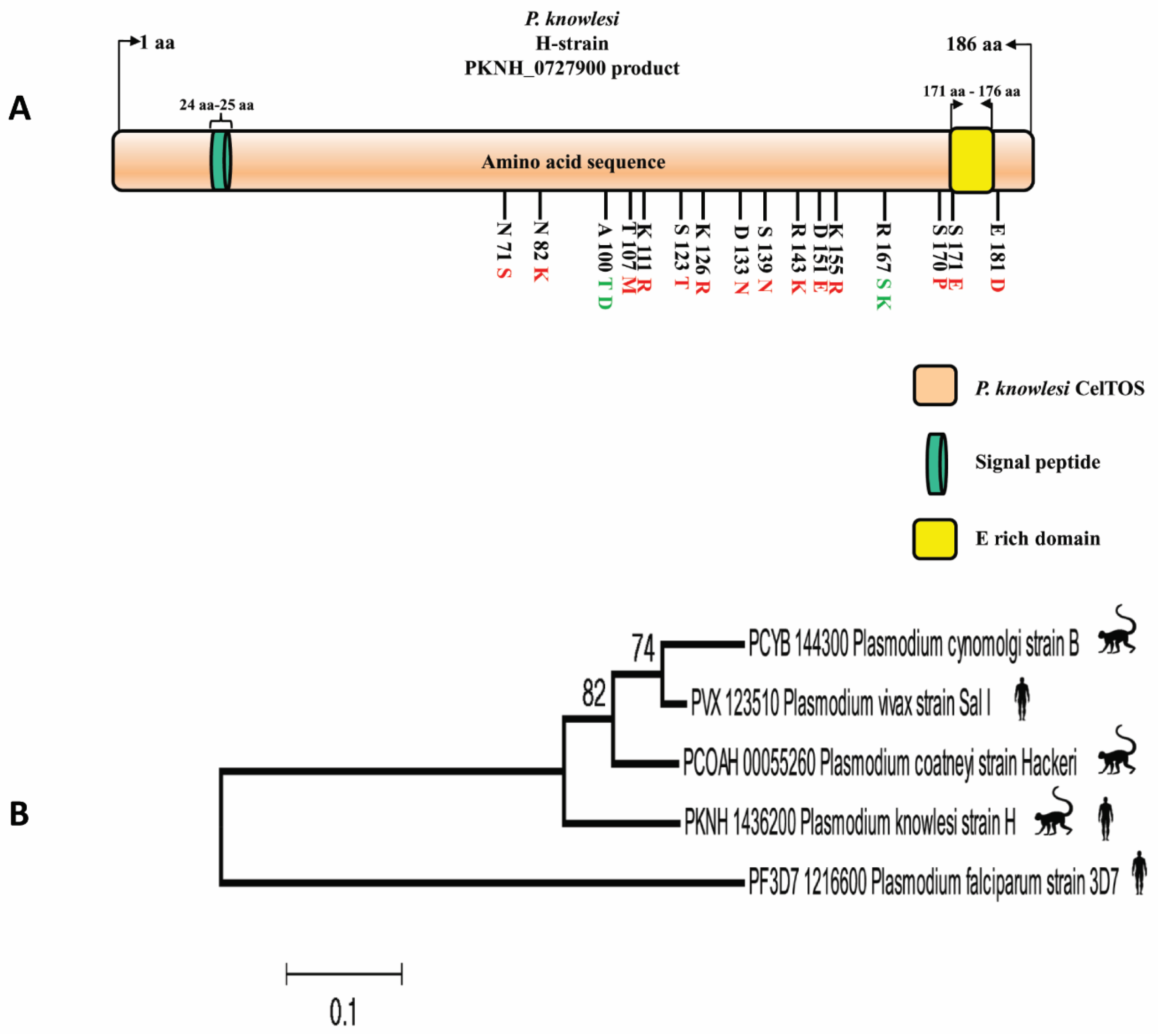
2.1. Sequence Information of PkCelTOS
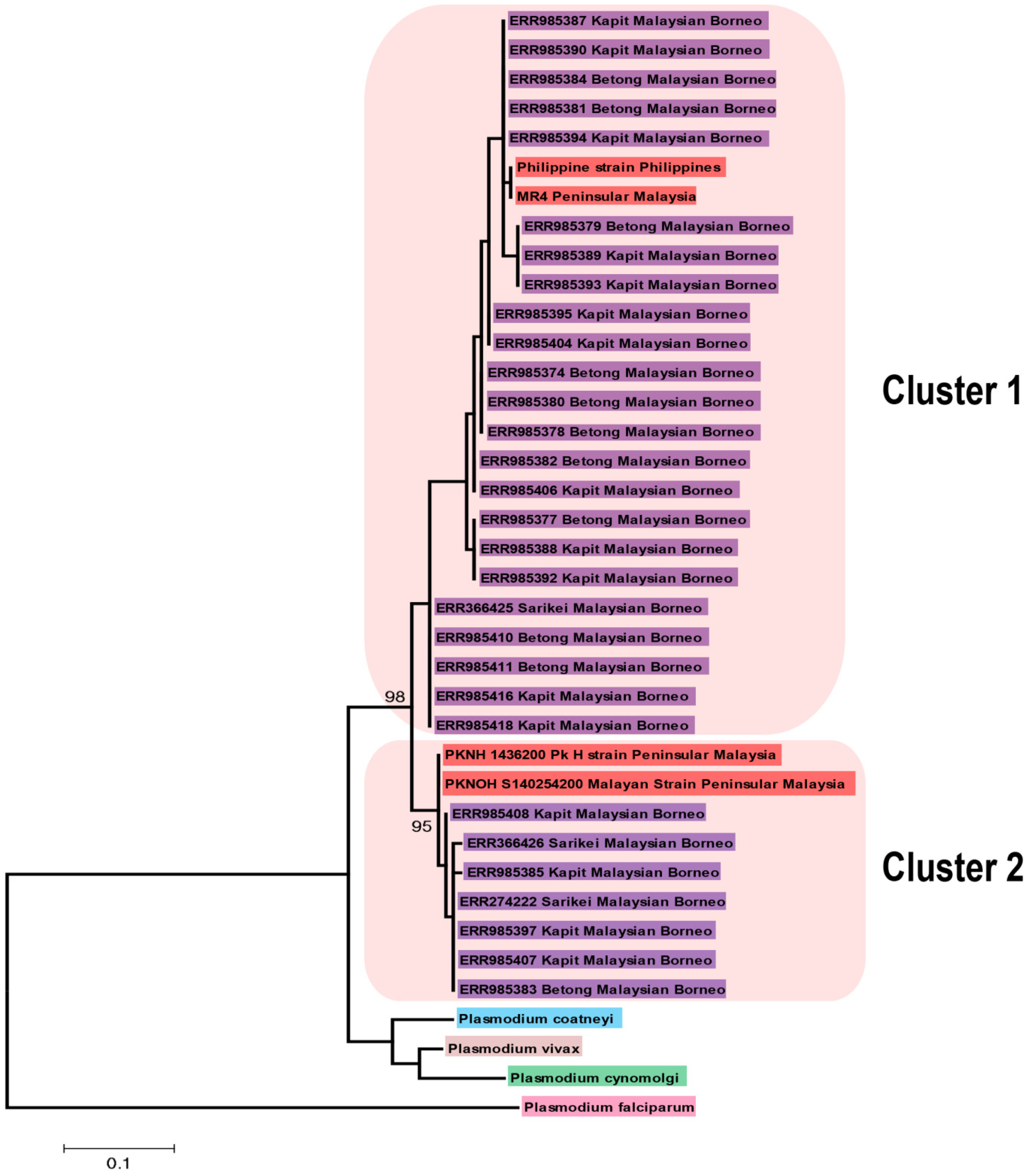
2.2. Phylogenetic Analysis
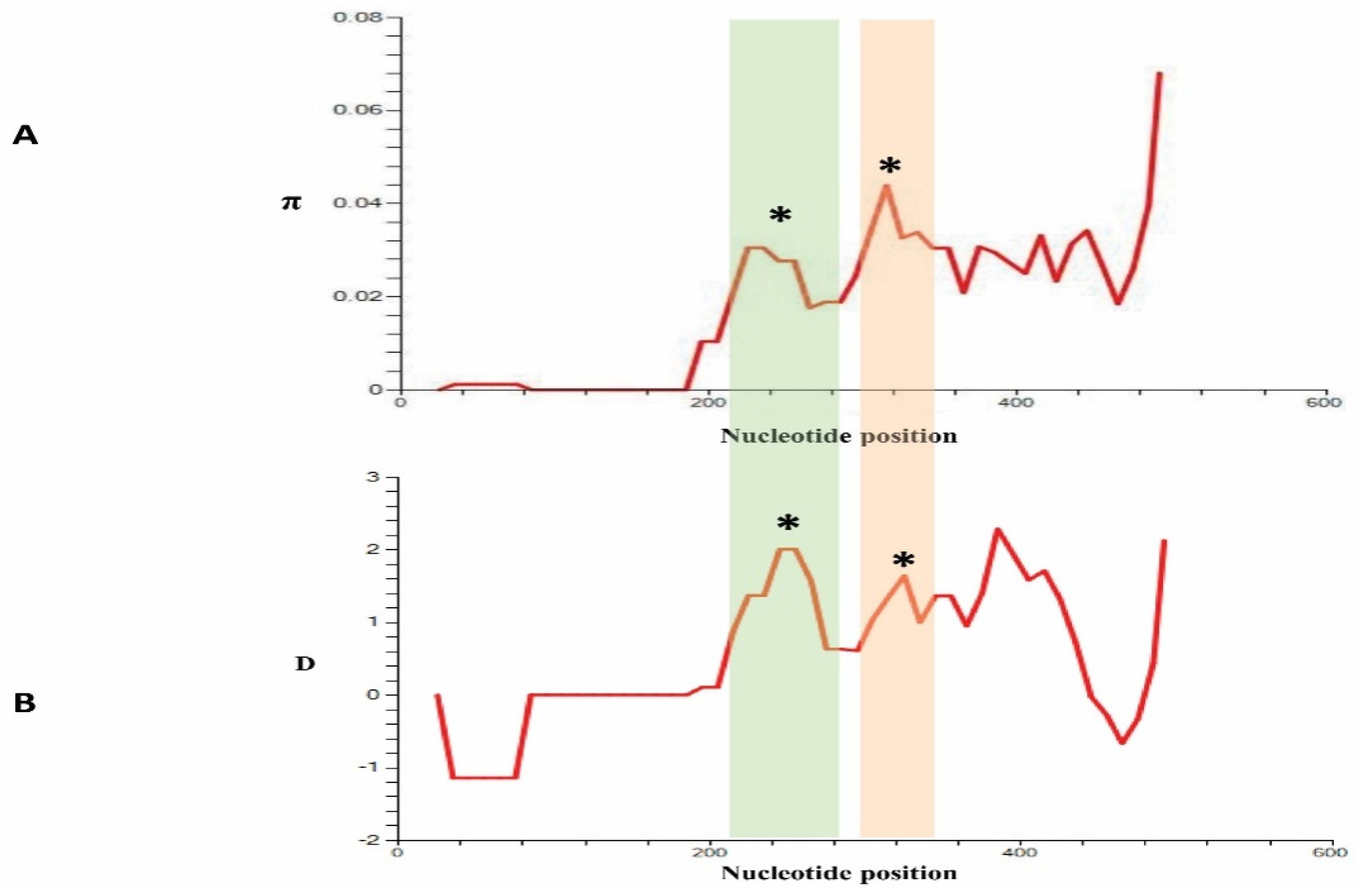
2.3. Sequence Diversity and Polymorphism
2.4. Natural Selection Test
2.5. Codon-Based Test Using Datamonkey Web Server for Natural Selection
2.6. Epitope Prediction
3. Results
3.1. PkCelTOS Sequence Identity among Ortholog Members
3.2. Phylogenetic Analysis of P. knowlesi CelTOS and Its Orthologs in Other Plasmodium Species
3.3. Nucleotide Diversity and Polymorphism of PkCelTOS in Clinical Samples
3.4. Natural Selection in PkCelTOS
3.5. Codon-Wise Analysis
3.6. B Cell Epitopes in PkCelTOS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 30 March 2023).
- World Health Organization. World Malaria Report; World Health Organisation: Geneva, Switzerland, 2022. [Google Scholar]
- Oguike, M.C.; Betson, M.; Burke, M.; Nolder, D.; Stothard, J.R.; Kleinschmidt, I.; Proietti, C.; Bousema, T.; Ndounga, M.; Tanabe, K.; et al. Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in African communities. Int. J. Parasitol. 2011, 41, 677–683. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Cox-Singh, J. Plasmodium knowlesi—An emerging pathogen. ISBT Sci. Ser. 2015, 10, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.J.; Tanomsing, N.; Nolder, D.; Oguike, M.; Jennison, C.; Pukrittayakamee, S.; Dolecek, C.; Hien, T.T.; do Rosario, V.E.; Arez, A.P.; et al. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J. Infect. Dis. 2010, 201, 1544–1550. [Google Scholar] [CrossRef]
- Tazi, L.; Ayala, F.J. Unresolved direction of host transfer of Plasmodium vivax v. P. simium and P. malariae v. P. brasilianum. Infect. Genet. Evol. 2011, 11, 209–221. [Google Scholar]
- Ta, T.H.; Hisam, S.; Lanza, M.; Jiram, A.I.; Ismail, N.; Rubio, J.M. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar. J. 2014, 13, 68. [Google Scholar] [CrossRef]
- Bogitsh, B.; Carter, C.; Oeltmann, T. Chapter 7—Blood and Tissue Protistans II: Human Malaria. In Human Parasitology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 5, pp. 111–133. [Google Scholar]
- White, N.J. Plasmodium knowlesi: The fifth human malaria parasite. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Zaidi, R.H.; Deshmukh, G.Y.; Saif, A.; Alshahrani, M.A.; Salam, S.S.; Elfaki, M.M.A.; Han, J.H.; Patgiri, S.J.; Quan, F.S. Genetic Diversity and Population Genetic Structure Analysis of Plasmodium knowlesi Thrombospondin-Related Apical Merozoite Protein (TRAMP) in Clinical Samples. Genes 2022, 13, 1944. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Deshmukh, G.Y.; Zaidi, R.H.; Saif, A.; Alshahrani, M.A.; Wazid, S.W.; Patgiri, S.J.; Quan, F.S. Identification, Mapping, and Genetic Diversity of Novel Conserved Cross-Species Epitopes of RhopH2 in Plasmodium knowlesi with Plasmodium vivax. Front. Cell. Infect. Microbiol. 2021, 11, 810398. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Quan, F.S. Plasmodium knowlesi clinical isolates from Malaysia show extensive diversity and strong differential selection pressure at the merozoite surface protein 7D (MSP7D). Malar. J. 2019, 18, 150. [Google Scholar] [CrossRef]
- Singh, B.; Kim Sung, L.; Matusop, A.; Radhakrishnan, A.; Shamsul, S.S.; Cox-Singh, J.; Thomas, A.; Conway, D.J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004, 363, 1017–1024. [Google Scholar] [CrossRef]
- Cox-Singh, J.; Davis, T.M.; Lee, K.S.; Shamsul, S.S.; Matusop, A.; Ratnam, S.; Rahman, H.A.; Conway, D.J.; Singh, B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 165–171. [Google Scholar] [CrossRef]
- Jeyaprakasam, N.K.; Liew, J.W.K.; Low, V.L.; Wan-Sulaiman, W.-Y.; Vythilingam, I. Plasmodium knowlesi infecting humans in Southeast Asia: What’s next? PLoS Neglected Trop. Dis. 2020, 14, e0008900. [Google Scholar] [CrossRef] [PubMed]
- Wharton, R.H.; Eyles, D.E. Anopheles hackeri, a vector of Plasmodium knowlesi in Malaya. Science 1961, 134, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Lau, Y.L.; Quan, F.S. Diversity and natural selection on the thrombospondin-related adhesive protein (TRAP) gene of Plasmodium knowlesi in Malaysia. Malar. J. 2018, 17, 274. [Google Scholar] [CrossRef]
- Assefa, S.; Lim, C.; Preston, M.D.; Duffy, C.W.; Nair, M.B.; Adroub, S.A.; Kadir, K.A.; Goldberg, J.M.; Neafsey, D.E.; Divis, P.; et al. Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi. Proc. Natl. Acad. Sci. USA 2015, 112, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.; Laing, A.; Warren, M.; Sandosham, A.; Wharton, R. Malaria parasites of the Malayan leaf monkeys of the genus Presbytis. Med J Malaya 1962, 17, 85–86. [Google Scholar]
- William, T.; Menon, J.; Rajahram, G.; Chan, L.; Ma, G.; Donaldson, S.; Khoo, S.; Frederick, C.; Jelip, J.; Anstey, N.M.; et al. Severe Plasmodium knowlesi malaria in a tertiary care hospital, Sabah, Malaysia. Emerg. Infect. Dis. 2011, 17, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.; Gupta, B.M.D. A Study of Monkey-Malaria, and Its Experimental Transmission to Man. Indian Med. Gaz. 1932, 67, 301–320. [Google Scholar]
- Singh, B.; Daneshvar, C. Human infections and detection of Plasmodium knowlesi. Clin. Microbiol. Rev. 2013, 26, 165–184. [Google Scholar] [CrossRef]
- Barber, B.E.; William, T.; Grigg, M.J.; Yeo, T.W.; Anstey, N.M. Limitations of microscopy to differentiate Plasmodium species in a region co-endemic for Plasmodium falciparum, Plasmodium vivax and Plasmodium knowlesi. Malar. J. 2013, 12, 8. [Google Scholar] [CrossRef]
- Vythilingam, I.; Wong, M.; Wan-Yussof, W. Current status of Plasmodium knowlesi vectors: A public health concern? Parasitology 2018, 145, 32–40. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Saif, A.; Quan, F.S. Diversity pattern of Plasmodium knowlesi merozoite surface protein 4 (MSP4) in natural population of Malaysia. PLoS ONE 2019, 14, e0224743. [Google Scholar] [CrossRef] [PubMed]
- Garzón-Ospina, D.; Buitrago, S.P.; Ramos, A.E.; Patarroyo, M.A. Identifying potential Plasmodium vivax sporozoite stage vaccine candidates: An analysis of genetic diversity and natural selection. Front. Genet. 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Sinnis, P.; Fidock, D.A. The RTS,S vaccine-a chance to regain the upper hand against malaria? Cell 2022, 185, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, D.E.; Juraska, M.; Bedford, T.; Benkeser, D.; Valim, C.; Griggs, A.; Lievens, M.; Abdulla, S.; Adjei, S.; Agbenyega, T. Genetic diversity and protective efficacy of the RTS, S/AS01 malaria vaccine. N. Engl. J. Med. 2015, 373, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Chu, K.B.; Quan, F.S. The Plasmodium knowlesi Pk41 surface protein diversity, natural selection, sub population and geographical clustering: A 6-cysteine protein family member. PeerJ 2018, 6, e6141. [Google Scholar] [CrossRef]
- Fong, M.Y.; Ahmed, M.A.; Wong, S.S.; Lau, Y.L.; Sitam, F. Genetic diversity and natural selection of the Plasmodium knowlesi circumsporozoite protein nonrepeat regions. PLoS ONE 2015, 10, e0137734. [Google Scholar] [CrossRef]
- Arevalo-Pinzon, G.; Garzon-Ospina, D.; Pulido, F.A.; Bermudez, M.; Forero-Rodriguez, J.; Rodriguez-Mesa, X.M.; Reyes-Guarin, L.P.; Suarez, C.F.; Patarroyo, M.A. Plasmodium vivax Cell Traversal Protein for Ookinetes and Sporozoites (CelTOS) Functionally Restricted Regions Are Involved in Specific Host-Pathogen Interactions. Front. Cell. Infect. Microbiol. 2020, 10, 119. [Google Scholar] [CrossRef]
- Pirahmadi, S.; Zakeri, S.; Mehrizi, A.A.; Djadid, N.D. Analysis of genetic diversity and population structure of gene encoding cell-traversal protein for ookinetes and sporozoites (CelTOS) vaccine candidate antigen in global Plasmodium falciparum populations. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 59, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt Chaves, L.; Perce-da-Silva, D.S.; Rodrigues-da-Silva, R.N.; Martins da Silva, J.H.; Cassiano, G.C.; Machado, R.L.; Pratt-Riccio, L.R.; Banic, D.M.; Lima-Junior, J.D. Plasmodium vivax Cell Traversal Protein for Ookinetes and Sporozoites (PvCelTOS) gene sequence and potential epitopes are highly conserved among isolates from different regions of Brazilian Amazon. PLoS Negl. Trop. Dis. 2017, 11, e0005344. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.; Salman, A.M.; Leoratti, F.; Lopez-Camacho, C.; Viveros-Sandoval, M.E.; Lall, A.; El-Turabi, A.; Bachmann, M.F.; Hill, A.V.; Janse, C.J.; et al. Evaluation of Plasmodium vivax Cell-Traversal Protein for Ookinetes and Sporozoites as a Preerythrocytic P. vivax Vaccine. Clin. Vaccine Immunol. CVI 2017, 24, e00501-16. [Google Scholar] [CrossRef]
- Kariu, T.; Ishino, T.; Yano, K.; Chinzei, Y.; Yuda, M. CelTOS, a novel malarial protein that mediates transmission to mosquito and vertebrate hosts. Mol. Microbiol. 2006, 59, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Jimah, J.R.; Salinas, N.D.; Sala-Rabanal, M.; Jones, N.G.; Sibley, L.D.; Nichols, C.G.; Schlesinger, P.H.; Tolia, N.H. Malaria parasite CelTOS targets the inner leaflet of cell membranes for pore-dependent disruption. eLife 2016, 5, e20621. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Mease, R.M.; De La Vega, P.; Savranskaya, T.; Polhemus, M.; Ockenhouse, C.; Angov, E. Immunization with pre-erythrocytic antigen CelTOS from Plasmodium falciparum elicits cross-species protection against heterologous challenge with Plasmodium berghei. PLoS ONE 2010, 5, e12294. [Google Scholar] [CrossRef]
- Bergmann-Leitner, E.S.; Legler, P.M.; Savranskaya, T.; Ockenhouse, C.F.; Angov, E. Cellular and humoral immune effector mechanisms required for sterile protection against sporozoite challenge induced with the novel malaria vaccine candidate CelTOS. Vaccine 2011, 29, 5940–5949. [Google Scholar] [CrossRef] [PubMed]
- Anum, D.; Kusi, K.A.; Ganeshan, H.; Hollingdale, M.R.; Ofori, M.F.; Koram, K.A.; Gyan, B.A.; Adu-Amankwah, S.; Badji, E.; Huang, J. Measuring naturally acquired ex vivo IFN-γ responses to Plasmodium falciparum cell-traversal protein for ookinetes and sporozoites (CelTOS) in Ghanaian adults. Malar. J. 2015, 14, 20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Espinosa, D.A.; Vega-Rodriguez, J.; Flores-Garcia, Y.; Noe, A.R.; Munoz, C.; Coleman, R.; Bruck, T.; Haney, K.; Stevens, A.; Retallack, D.; et al. The Plasmodium falciparum Cell-Traversal Protein for Ookinetes and Sporozoites as a Candidate for Preerythrocytic and Transmission-Blocking Vaccines. Infect. Immun. 2017, 85, 10–1128. [Google Scholar] [CrossRef]
- Trial of a Falciparum Malaria Protein (FMP012), E. Coli-expressed PfCelTOS, in Healthy Malaria-Naive Adults. ClinicalTrials.Gov. 2021. Available online: https://www.clinicaltrials.gov/study/NCT01540474?cond=Trial%20of%20a%20Falciparum%20Malaria%20Protein%20(FMP012),%20E.%20Coli-expressed%20PfCelTOS,%20in%20Healthy%20Malaria-Naive%20Adults&rank=1 (accessed on 20 March 2023).
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of Hepatitis A Virus-Neutralizing Antibody by a Virus-Specific Synthetic Peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Janin, J.; Wodak, S. Conformation of amino acid side-chains in proteins. J. Mol. Biol. 1978, 125, 357–386. [Google Scholar] [CrossRef]
- Muh, F.; Kim, N.; Nyunt, M.H.; Firdaus, E.R.; Han, J.-H.; Hoque, M.R.; Lee, S.-K.; Park, J.-H.; Moon, R.W.; Lau, Y.L. Cross-species reactivity of antibodies against Plasmodium vivax blood-stage antigens to Plasmodium knowlesi. PLoS Neglected Trop. Dis. 2020, 14, e0008323. [Google Scholar] [CrossRef]
- Longley, R.J.; Grigg, M.J.; Schoffer, K.; Obadia, T.; Hyslop, S.; Piera, K.A.; Nekkab, N.; Mazhari, R.; Takashima, E.; Tsuboi, T. Plasmodium vivax malaria serological exposure markers: Assessing the degree and implications of cross-reactivity with P. knowlesi. Cell Rep. Med. 2022, 3, 100662. [Google Scholar] [CrossRef]
- Mota, M.M.; Pradel, G.; Vanderberg, J.P.; Hafalla, J.C.; Frevert, U.; Nussenzweig, R.S.; Nussenzweig, V.; Rodriguez, A. Migration of Plasmodium sporozoites through cells before infection. Science 2001, 291, 141–144. [Google Scholar] [CrossRef]
- Vanderberg, J.P.; Chew, S.; Stewart, M.J. Plasmodium sporozoite interactions with macrophages in vitro: A videomicroscopic analysis. J. Protozool. 1990, 37, 528–536. [Google Scholar] [CrossRef]
- Amino, R.; Giovannini, D.; Thiberge, S.; Gueirard, P.; Boisson, B.; Dubremetz, J.F.; Prevost, M.C.; Ishino, T.; Yuda, M.; Menard, R. Host cell traversal is important for progression of the malaria parasite through the dermis to the liver. Cell Host Microbe 2008, 3, 88–96. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Fauzi, M.; Han, E.T. Genetic diversity and natural selection of Plasmodium knowlesi merozoite surface protein 1 paralog gene in Malaysia. Malar. J. 2018, 17, 115. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Fong, M.Y.; Lau, Y.L.; Yusof, R. Clustering and genetic differentiation of the normocyte binding protein (nbpxa) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia and Malaysia Borneo. Malar. J. 2016, 15, 241. [Google Scholar] [CrossRef]
- Pinheiro, M.M.; Ahmed, M.A.; Millar, S.B.; Sanderson, T.; Otto, T.D.; Lu, W.C.; Krishna, S.; Rayner, J.C.; Cox-Singh, J. Plasmodium knowlesi genome sequences from clinical isolates reveal extensive genomic dimorphism. PLoS ONE 2015, 10, e0121303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | No. Samples | SNPs | Syn | Non-Syn | No. Haplotype | Diversity ± SD | Taj D | Fu and Li’s F* | Fu and Li’s D* | |
|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Nucleotide | |||||||||
| PkCelTOS | 34 | 28 | 12 | 16 | 17 | 0.954 ± 0.016 | 0.02111 ± 0.00105 | 1.52705 p > 0.10 | 1.10034 p > 0.10 | 0.63460 p > 0.10 |
| CelTOS | Polymorphic Changes Observed in P. knowlesi | Fixed Differences between Species | Neutrality Index | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pk vs. Pco | Pk vs. Pcy | Pk vs. Pv | Pk vs. Pco | Pk vs. Pcy | Pk vs. Pv | ||||||
| Syn | Non-Syn | Syn | NonSyn | Syn | Non-Syn | Syn | Non-Syn | ||||
| Full length | 12 | 16 | 15 | 22 | 20 | 32 | 21 | 20 | 0.909 | 0.909 | 1.400 |
| IEDB Server | Bcpred Server | |||||||
|---|---|---|---|---|---|---|---|---|
| No. | Start AA | End AA | Peptide | Length | Start AA | End AA | Peptide | Length |
| 1 | 96 | 102 | AQLKATA | 7 | 125 | 133 | IKPPRIKED | 9 |
| 2 | 124 | 133 | TIKPPRIKED | 10 | - | - | - | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.A.; Baruah, P.; Saif, A.; Han, J.-H.; Al-Zharani, M.; Wazid, S.W.; Alkahtani, S.; Patgiri, S.J.; Al-Eissa, M.S.; Quan, F.-S. In Silico Analysis Reveals High Levels of Genetic Diversity of Plasmodium knowlesi Cell Traversal Protein for Ookinetes and Sporozoites (PkCelTOS) in Clinical Samples. Trop. Med. Infect. Dis. 2023, 8, 380. https://doi.org/10.3390/tropicalmed8080380
Ahmed MA, Baruah P, Saif A, Han J-H, Al-Zharani M, Wazid SW, Alkahtani S, Patgiri SJ, Al-Eissa MS, Quan F-S. In Silico Analysis Reveals High Levels of Genetic Diversity of Plasmodium knowlesi Cell Traversal Protein for Ookinetes and Sporozoites (PkCelTOS) in Clinical Samples. Tropical Medicine and Infectious Disease. 2023; 8(8):380. https://doi.org/10.3390/tropicalmed8080380
Chicago/Turabian StyleAhmed, Md Atique, Pratisthita Baruah, Ahmed Saif, Jin-Hee Han, Mohammed Al-Zharani, Syeda Wasfeea Wazid, Saad Alkahtani, Saurav J. Patgiri, Mohammed S. Al-Eissa, and Fu-Shi Quan. 2023. "In Silico Analysis Reveals High Levels of Genetic Diversity of Plasmodium knowlesi Cell Traversal Protein for Ookinetes and Sporozoites (PkCelTOS) in Clinical Samples" Tropical Medicine and Infectious Disease 8, no. 8: 380. https://doi.org/10.3390/tropicalmed8080380
APA StyleAhmed, M. A., Baruah, P., Saif, A., Han, J.-H., Al-Zharani, M., Wazid, S. W., Alkahtani, S., Patgiri, S. J., Al-Eissa, M. S., & Quan, F.-S. (2023). In Silico Analysis Reveals High Levels of Genetic Diversity of Plasmodium knowlesi Cell Traversal Protein for Ookinetes and Sporozoites (PkCelTOS) in Clinical Samples. Tropical Medicine and Infectious Disease, 8(8), 380. https://doi.org/10.3390/tropicalmed8080380