
Analysis of Puumala orthohantavirus Genome Variants Identified in the Territories of Volga Federal District

 ,
, 

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khismatullina, N.A.; Karimov, M.M.; Khaertynov, K.S.; Shuralev, E.A.; Morzunov, S.P.; Khaertynova, I.M.; Ivanov, A.A.; Milova, I.V.; Khakimzyanova, M.B.; Sayfullina, G.S.; et al. Epidemiological dynamics of nephropathia epidemica in the Republic of Tatarstan, Russia, during the period of 1997–2013. Epidemiol. Infect. 2016, 144, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Onishchenko, G.G.; Ezhlova, E.B. Epidemiologic surveillance and prophylaxis of hemorrhagic fever with renal syndrome in Russian Federation. J. Microbiol. Epidemiol. Immunobiol. 2013, 4, 23–32. [Google Scholar]
- Matrosov, A.N.; Korneev, M.G.; Chekashov, V.N.; Ivanova, A.V.; Sludsky, A.A.; Zakharov, K.S.; Maharramov, S.V.; Selenina, A.G.; Shilov, M.M.; Popov, N.V. An Overview of the Number of Carriers and Vectors of Zoonoses, the Epizootic and Epidemiological Situation in the Volga Federal District in the Second Half of 2020 and the Forecast for the First Half of 2021. Available online: microbe.ru/files/PFO_rev2020_II_prog2021_I.pdf (accessed on 12 December 2021). (In Russian).
- Cunze, S.; Kochmann, J.; Kuhn, T.; Frank, R.; Dörge, D.D.; Klimpel, S. Spatial and temporal patterns of human Puumala virus (PUUV) infections in Germany. PeerJ 2018, 6, e4255. [Google Scholar] [CrossRef] [Green Version]
- Watson, D.; Sargianou, M.; Papa, A.; Chra, P.; Starakis, I.; Panos, G. Epidemiology of Hantavirus infections in humans: A comprehensive, global overview. Crit. Rev. Microbiol. 2014, 40, 261–272. [Google Scholar] [CrossRef]
- Ettinger, J.; Hofmann, J.; Enders, M.; Tewald, F.; Oehme, R.M.; Rosenfeld, U.M.; Ali, H.S.; Schlegel, M.; Essbauer, S.; Osterberg, A.; et al. Multiple Synchronous Outbreaks of Puumala Virus, Germany, 2010. Emerg. Infect. Dis. 2012, 18, 1461–1464. [Google Scholar] [CrossRef]
- Khaiboullina, S.; Morzunov, S.P.; Jeor, S.C.S. Hantaviruses: Molecular Biology, Evolution and Pathogenesis. Curr. Mol. Med. 2005, 5, 773–790. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Figueiredo, L.T.M.; Vapalahti, O. A Global Perspective on Hantavirus Ecology, Epidemiology, and Disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef]
- Schwarz, A.C.; Ranft, U.; Piechotowski, I.; Childs, J.E.; Brockmann, S.O. Risk Factors for Human Infection with Puumala Virus, Southwestern Germany. Emerg. Infect. Dis. 2009, 15, 1032–1039. [Google Scholar] [CrossRef]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of Complete Puumala Virus Genome, Finland. Emerg. Infect. Dis. 2012, 18, 2070–2072. [Google Scholar] [CrossRef]
- Ali, H.S.; Drewes, S.; de Melo, V.W.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef]
- Sundström, K.B.; Stoltz, M.; Lagerqvist, N.; Lundkvist, Å.; Nemirov, K.; Klingström, J. Characterization of Two Substrains of Puumala Virus That Show Phenotypes That Are Different from Each Other and from the Original Strain. J. Virol. 2010, 85, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Županc, T.A.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [Green Version]
- De Vries, A.; Vennema, H.; Bekker, D.L.; Maas, M.; Adema, J.; Opsteegh, M.; Van Der Giessen, J.W.B.; Reusken, C.B.E.M. Characterization of Puumala hantavirus in bank voles from two regions in the Netherlands where human cases occurred. J. Gen. Virol. 2016, 97, 1500–1510. [Google Scholar] [CrossRef]
- Reuter, M.; Krüger, D.H. The nucleocapsid protein of hantaviruses: Much more than a genome-wrapping protein. Virus Genes 2018, 54, 5–16. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90 Pt 8, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Nemirov, K.; Leirs, H.; Lundkvist, Å.; Olsson, G.E. Puumala hantavirus and Myodes glareolus in northern Europe: No evidence of co-divergence between genetic lineages of virus and host. J. Gen. Virol. 2010, 91 Pt 5, 1262–1274. [Google Scholar] [CrossRef]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular Evolution of Puumala Hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef]
- Davidyuk, Y.; Shamsutdinov, A.; Kabwe, E.; Ismagilova, R.; Martynova, E.; Belyaev, A.; Shuralev, E.; Trifonov, V.; Savitskaya, T.; Isaeva, G.; et al. Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens 2020, 9, 540. [Google Scholar] [CrossRef]
- Davidyuk, Y.N.; Kabwe, E.; Shamsutdinov, A.F.; Knyazeva, A.V.; Martynova, E.V.; Ismagilova, R.K.; Trifonov, V.A.; Savitskaya, T.A.; Isaeva, G.S.; Urbanowicz, R.A.; et al. The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan. Pathogens 2021, 10, 1169. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef] [Green Version]
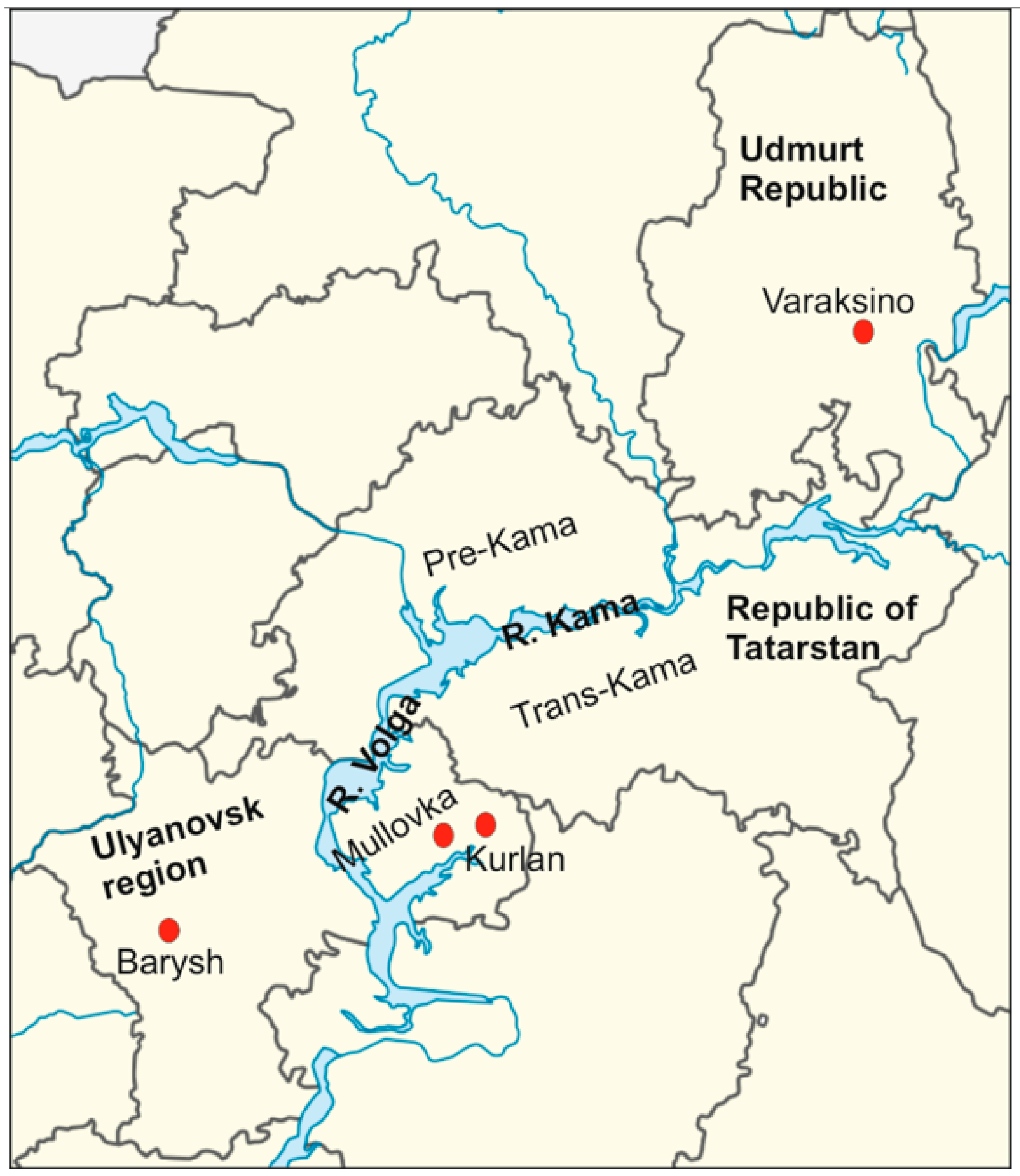
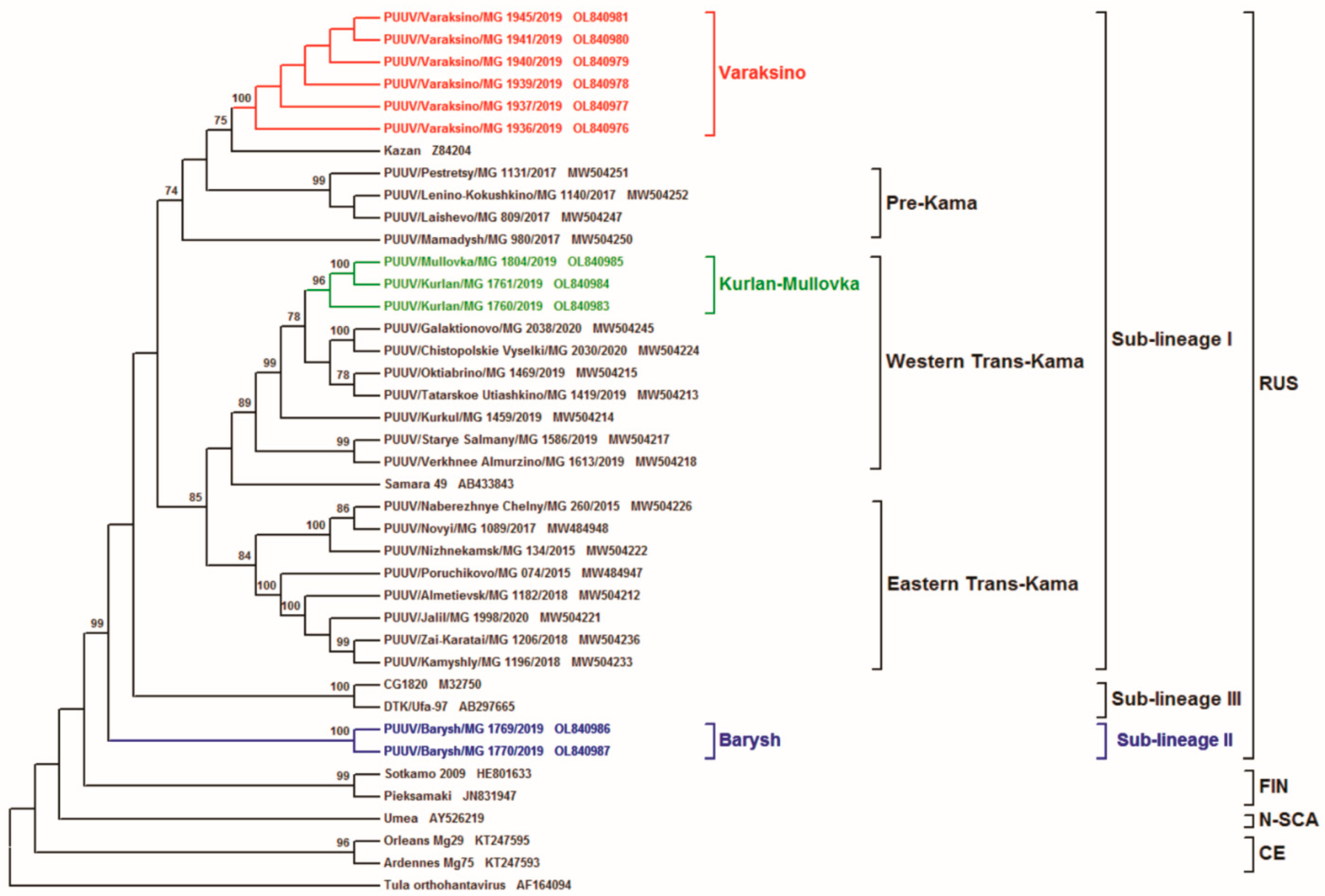
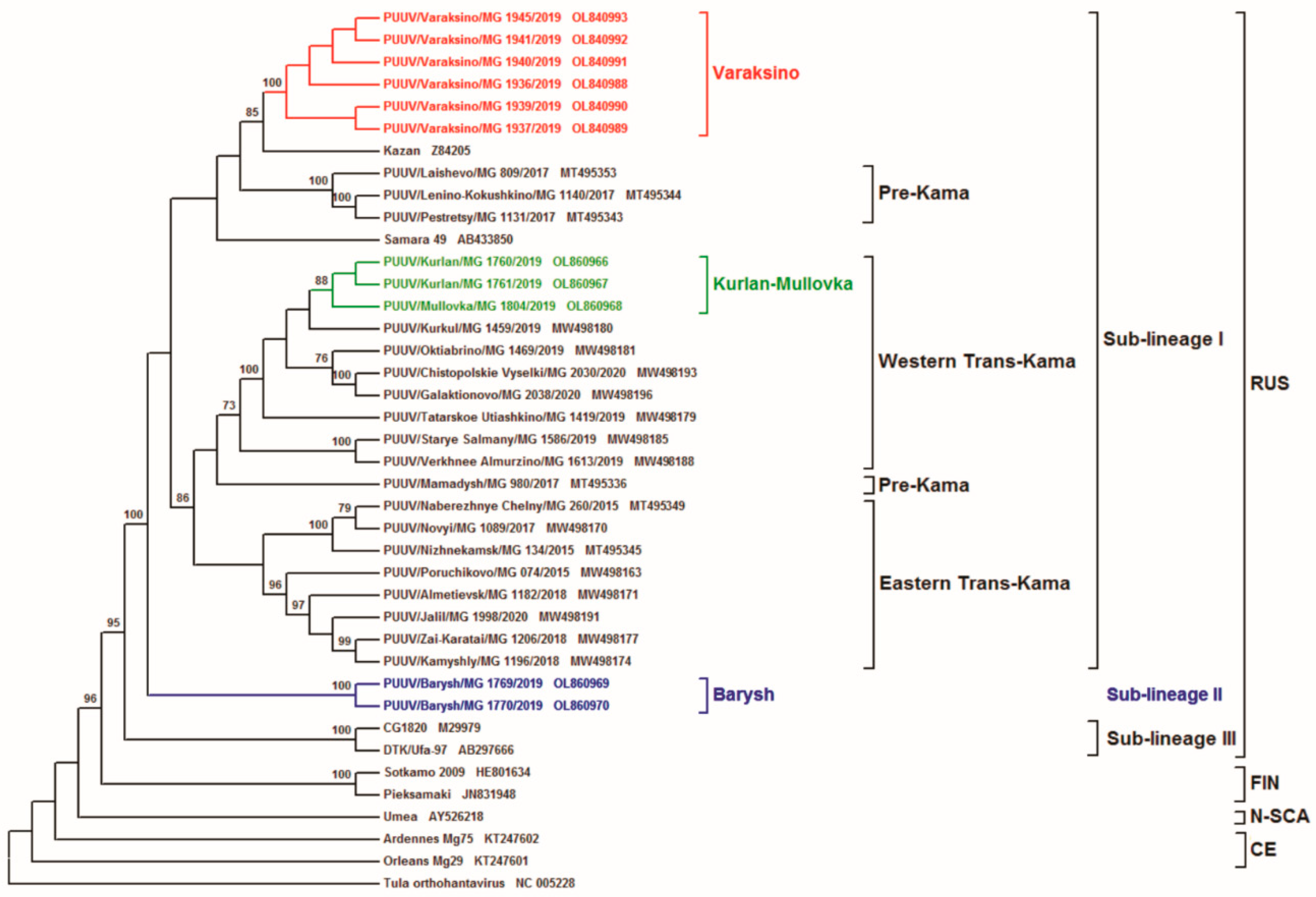
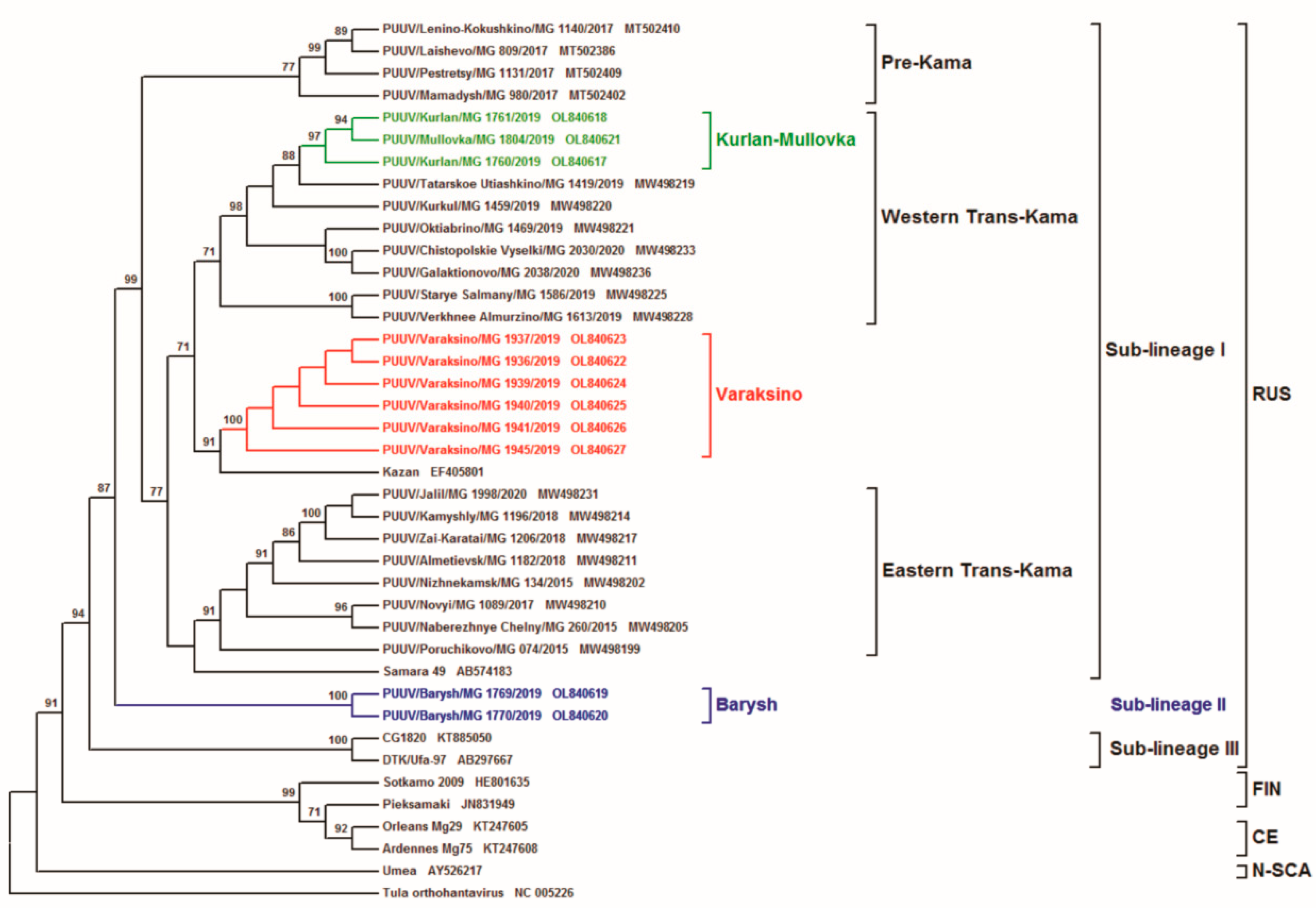

{kind=link}
{kind=link}
{kind=link}
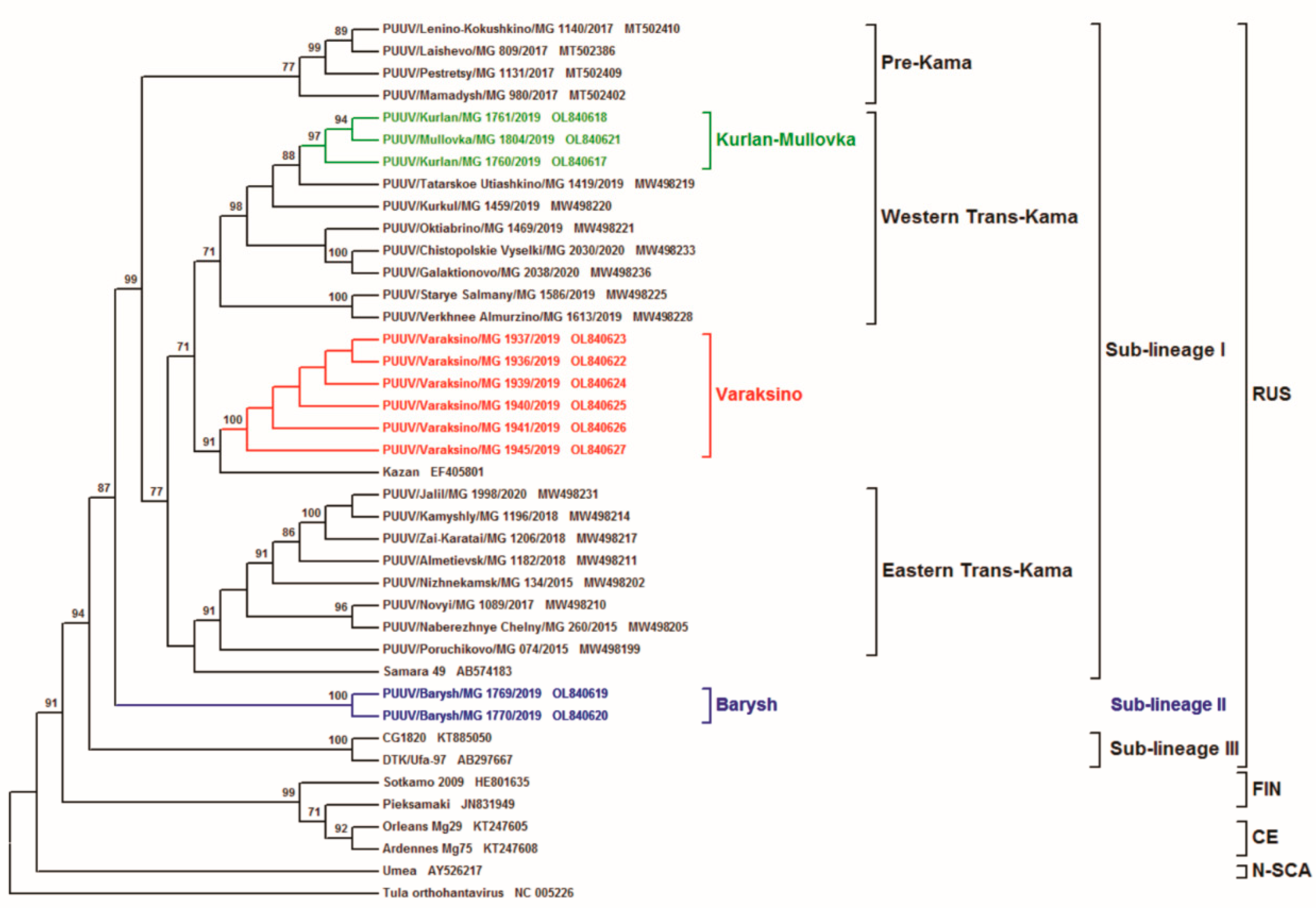{kind=link}
| Strain | Location in the RT | Abbreviation | GenBank Accession Number | ||
|---|---|---|---|---|---|
| S Segment | M Segment | L Segment | |||
| PUUV/Poruchikovo/MG_074/2015 | Eastern Trans-Kama | MG074 | MW484947 | MW498163 | MW498199 |
| PUUV/Nizhnekamsk/MG_134/2015 | Eastern Trans-Kama | MG134 | MW504222 | MT495345 | MW498202 |
| PUUV/Naberezhnye Chelny/MG_260/2015 | Eastern Trans-Kama | MG260 | MW504226 | MT495349 | MW498205 |
| PUUV/Laishevo/MG_809/2017 | Pre-Kama | MG809 | MW504247 | MT495353 | MT502386 |
| PUUV/Mamadysh/MG_980/2017 | Pre-Kama | MG980 | MW504250 | MT495336 | MT502402 |
| PUUV/Novyi/MG_1089/2017 | Eastern Trans-Kama | MG1089 | MW484948 | MW498170 | MW498210 |
| PUUV/Pestretsy/MG_1131/2017 | Pre-Kama | MG1131 | MW504251 | MT495343 | MT502409 |
| PUUV/Lenino-Kokushkino/MG_1140/2017 | Pre-Kama | MG1140 | MW504252 | MT495344 | MT502410 |
| PUUV/Almetievsk/MG_1182/2018 | Eastern Trans-Kama | MG1182 | MW504212 | MW498171 | MW498211 |
| PUUV/Zai-Karatai/MG_1206/2018 | Eastern Trans-Kama | MG1206 | MW504236 | MW498177 | MW498217 |
| PUUV/Kamyshly/MG_1196/2018/2018 | Eastern Trans-Kama | MG1196 | MW504233 | MW498174 | MW498214 |
| PUUV/Tatarskoe Utiashkino/MG_1419/2019 | Western Trans-Kama | MG1419 | MW504213 | MW498179 | MW498219 |
| PUUV/Kurkul/MG_1459/2019 | Western Trans-Kama | MG1459 | MW504214 | MW498180 | MW498220 |
| PUUV/Oktiabrino/MG_1469/2019 | Western Trans-Kama | MG1469 | MW504215 | MW498181 | MW498221 |
| PUUV/Starye Salmany/MG_1586/2019 | Western Trans-Kama | MG1586 | MW504217 | MW498185 | MW498225 |
| PUUV/Dzhalil/MG_1998/2020 | Eastern Trans-Kama | MG1998 | MW504221 | MW498191 | MW498231 |
| PUUV/Verkhnee Almurzino/MG_1613/2019 | Western Trans-Kama | MG1613 | MW504218 | MW498188 | MW498228 |
| PUUV/Chistopolskie Vyselki/MG_2030/2020 | Western Trans-Kama | MG2030 | MW504224 | MW498193 | MW498233 |
| PUUV/Galaktionovo/MG_2038/2020 | Western Trans-Kama | MG2038 | MW504245 | MW498196 | MW498236 |
| Puu/Kazan | «Kazan» | Z84204 | Z84205 | EF405801 | |
| Samara_49/CG/2005 | «Samara» | AB433843 | AB433850 | AB574183 | |
| CG1820 | CG1820 | M32750 | M29979 | KT885050 | |
| DTK/Ufa-97 | «Ufa» | AB297665 | AB297666 | AB297667 | |
| Sotkamo 2009 | «Sotkamo» | HE801633 | HE801634 | HE801635 | |
| PUUV/Pieksamaki/human_lung/2008 | «Pieksamaki» | JN831947 | JN831948 | JN831949 | |
| Umea/hu | «Umea» | AY526219 | AY526218 | AY526217 | |
| PUUV/Orleans/Mg29/2010 | «Orleans» | KT247595 | KT247601 | KT247605 | |
| PUUV/Ardennes/Mg75/2011 | «Ardennes» | KT247593 | KT247602 | KT247608 | |
| Tula orthohantavirus | «Tula» | AF164094 | NC_005228 | NC_005226 | |
| PUUV Strain | Identity, % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| S Segment | M Segment | L Segment | |||||||
| Udmurt Republic | Ulyanovsk Region | Udmurt Republic | Ulyanovsk Region | Udmurt Republic | Ulyanovsk Region | ||||
| Varaksino | Kurlan–Mullovka | Barysh | Varaksino | Kurlan–Mullovka | Barysh | Varaksino | Kurlan–Mullovka | Barysh | |
| Varaksino | 100.0 | 94.2–95.1 | 93.8–93.9 | 99.9–100.0 | 93.7–94.5 | 92.9–93.0 | 100.0 | 93.0–93.7 | 85.9 |
| Kurlan–Mullovka | 98.5–99.5 | 93.2–93.7 | 99.0–99.7 | 91.6–92.1 | 98.0–99.3 | 86.2–86.4 | |||
| Barysh | 99.8 | 100.0 | 100.0 | ||||||
| Location | RUS | FIN | CE | N-SCA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RT | Samara | Kazan | CG1820 | Ufa | Sotkamo | Pieksamaki | Ardennes | Orleans | Umea | |
| S segment | ||||||||||
| Varaksino | 94.1–96.2 | 94.9 | 96.5 | 94.3 | 94.4 | 84.9 | 86.2 | 83.2 | 83.3 | 83.6 |
| Kurlan–Mullovka | 94.2–98.5 | 96.1–96.4 | 94.5–94.9 | 93.8–94.3 | 93.9–94.4 | 85.3–85.5 | 86.3–86.6 | 83.3–84.1 | 83.0–83.2 | 84.6–84.8 |
| Barysh | 92.9–94.2 | 93.9–94.0 | 93.5 | 93.6–93.8 | 93.7–93.9 | 85.7–85.8 | 85.7 | 83.6 | 82.6 | 84.6 |
| M segment | ||||||||||
| Varaksino | 93.0–95.4 | 93.7–93.8 | 95.9–96.0 | 86.8–86.9 | 87.4–87.5 | 83.6–83.7 | 83.1–83.2 | 81.1–81.2 | 79.3–79.4 | 81.2–81.3 |
| Kurlan–Mullovka | 93.0–98.8 | 92.4–92.8 | 93.8–94.6 | 85.3 | 85.9 | 84.1–84.5 | 83.9–84.1 | 81.1–81.5 | 80.3–80.5 | 81.2–81.5 |
| Barysh | 90.6–93.3 | 91.5 | 91.9 | 86.0 | 86.6 | 83.2 | 82.5 | 81.3 | 80.2 | 81.0 |
| L segment | ||||||||||
| Varaksino | 92.7–95.0 | 93.7 | 95.5 | 85.9 | 85.9 | 83.1 | 83.7 | 81.6 | 82.8 | 79.9 |
| Kurlan–Mullovka | 91.7–97.7 | 93.0–93.4 | 92.7–93.7 | 85.4–86.1 | 85.4–86.1 | 82.3–82.6 | 82.9–83.1 | 81.8 | 82.6–82.9 | 79.6–79.9 |
| Barysh | 84.7–87.2 | 85.7 | 85.6 | 84.1 | 84.1 | 82.1 | 80.6 | 78.9 | 79.9 | 80.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabwe, E.; Al Sheikh, W.; Shamsutdinov, A.F.; Ismagilova, R.K.; Martynova, E.V.; Ohlopkova, O.V.; Yurchenko, Y.A.; Savitskaya, T.A.; Isaeva, G.S.; Khaiboullina, S.F.; et al. Analysis of Puumala orthohantavirus Genome Variants Identified in the Territories of Volga Federal District. Trop. Med. Infect. Dis. 2022, 7, 46. https://doi.org/10.3390/tropicalmed7030046
Kabwe E, Al Sheikh W, Shamsutdinov AF, Ismagilova RK, Martynova EV, Ohlopkova OV, Yurchenko YA, Savitskaya TA, Isaeva GS, Khaiboullina SF, et al. Analysis of Puumala orthohantavirus Genome Variants Identified in the Territories of Volga Federal District. Tropical Medicine and Infectious Disease. 2022; 7(3):46. https://doi.org/10.3390/tropicalmed7030046
Chicago/Turabian StyleKabwe, Emmanuel, Walaa Al Sheikh, Anton F. Shamsutdinov, Ruzilya K. Ismagilova, Ekaterina V. Martynova, Olesia V. Ohlopkova, Yuri A. Yurchenko, Tatiana A. Savitskaya, Guzel S. Isaeva, Svetlana F. Khaiboullina, and et al. 2022. "Analysis of Puumala orthohantavirus Genome Variants Identified in the Territories of Volga Federal District" Tropical Medicine and Infectious Disease 7, no. 3: 46. https://doi.org/10.3390/tropicalmed7030046
APA StyleKabwe, E., Al Sheikh, W., Shamsutdinov, A. F., Ismagilova, R. K., Martynova, E. V., Ohlopkova, O. V., Yurchenko, Y. A., Savitskaya, T. A., Isaeva, G. S., Khaiboullina, S. F., Rizvanov, A. A., Morzunov, S. P., & Davidyuk, Y. N. (2022). Analysis of Puumala orthohantavirus Genome Variants Identified in the Territories of Volga Federal District. Tropical Medicine and Infectious Disease, 7(3), 46. https://doi.org/10.3390/tropicalmed7030046