Abstract
Ticks can carry and transmit a large number of pathogens, including bacteria, viruses and protozoa, posing a huge threat to human health and animal husbandry. Previous investigations have shown that the dominant species of ticks in Shanghai are Haemaphysalis flava and Haemaphysalis longicornis. However, no relevant investigations and research have been carried out in recent decades. Therefore, we investigated the bacterial communities and tick-borne pathogens (TBPs) in Haemaphysalis spp. from Shanghai, China. Ixodid ticks were collected from 18 sites in Shanghai, China, and identified using morphological and molecular methods. The V3–V4 hypervariable regions of the bacterial 16S rRNA gene were amplified from the pooled tick DNA samples and subject to metagenomic analysis. The microbial diversity in the tick samples was estimated using the alpha diversity that includes the observed species index and Shannon index. The Unifrac distance matrix as determined using the QIIME software was used for unweighted Unifrac Principal coordinates analysis (PCoA). Individual tick DNA samples were screened with genus-specific or group-specific nested polymerase chain reaction (PCR) for these TBPs and combined with a sequencing assay to confirm the results of the V3–V4 hypervariable regions of the bacterial 16S rRNA gene. We found H. flava and H. longicornis to be the dominant species of ticks in Shanghai in this study. Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria are the main bacterial communities of Haemaphysalis spp. The total species abundances of Proteobacteria, Firmicutes and Bacteroidetes, are 48.8%, 20.8% and 18.1%, respectively. At the level of genus analysis, H. longicornis and H. flava carried at least 946 genera of bacteria. The bacteria with high abundance include Lactobacillus, Coxiella, Rickettsia and Muribaculaceae. Additionally, Rickettsia rickettsii, Rickettsia japonica, Candidatus Rickettsia jingxinensis, Anaplasma bovis, Ehrlichia ewingii, Ehrlichia chaffeensis, Coxiella spp. and Coxiella-like endosymbiont were detected in Haemaphysalis spp. from Shanghai, China. This study is the first report of bacterial communities and the prevalence of some main pathogens in Haemaphysalis spp. from Shanghai, China, and may provide insights and evidence for bacterial communities and the prevalence of the main pathogen in ticks. This study also indicates that people and other animals in Shanghai, China, are exposed to several TBPs.
1. Introduction
Ticks are the second largest infectious agent in the world after mosquitoes, with a wide variety of species and a wide range of animal hosts [1]. They can carry and transmit a large number of pathogens, including bacteria, viruses and protozoa, posing a huge threat to human health and animal husbandry [2]. Ticks are known to harbor a number of veterinary and medically important bacterial species within the Rickettsia, Anaplasma, Bartonella, Coxiella and Ehrlichia genera [2,3]. As most ticks exhibit two-host or three-host life cycles, they are capable of supporting the transmission of pathogens between hosts, in which humans frequently serve as accidental hosts [4]. Multiple pathogenic agents may also be carried by an individual tick, which could transmit these pathogens to the human hosts bitten by ticks [3,4]. The main tick-borne diseases reported in China are forest encephalitis, Crimean Congo hemorrhagic fever, Lyme disease, rabbit fever, Q fever, North Asian tick-borne spotted fever, rickettsiosis, ehrlichiosis, anaplasmosis, babesiosis and Taylor disease [5,6,7,8]. Ticks of Haemaphysalis spp. have been implicated as potential disease vectors to humans and animals worldwide [3,9]. Various pathogenic bacteria have been previously detected in Haemaphysalis spp., including the disease agents for rickettsia spotted fever, tick typhus, anaplasmosis and ehrlichiosis [3,10,11,12].
Traditional detection methods (PCR, culture and serological methods), which have played a very important role in previous pathogen detection and identification, cannot detect all pathogens, especially when in low abundance and with unknown pathogens. Additionally, traditional detection methods are extremely dependent on known pathogens. However, metagenomic sequencing can identify a large number of micro pathogens, including unknown pathogens, in tick microflora captured in the field and does not depend on known nucleic acid sequences [13,14,15]. This method can be used not only to monitor microbial communities in infectious insect vectors but also as an ideal tool for monitoring emerging tick-borne diseases [13,15]. It can also provide a more thorough understanding of the ecological factors related to the prevalence and persistence of the vector-related microbial pedigree, which will help to predict and prevent the spread of diseases [13,15,16,17].
More and more tick-borne diseases and pathogens have been discovered. Recently, a new species of Yezo virus of the genus Nairobi virus was discovered in ticks in Hokkaido, Japan, and caused multiple infections [18]. Since the early 1980s, 34 pathogens of tick-borne diseases have been identified in mainland China, including eight species of spotted fever group Rickettsia, seven species of Anaplasma, six species of Borrelia burgdorferi, 11 species of Babesia and severe fever with thrombocytopenia syndrome new Bunyavirus (SFTSV) and Alongshan Virus (ALSV) [7,19]. Tick-borne diseases and tick bites frequently occur in provinces and cities around Shanghai, China, which seriously endangers the life and health of local residents. Human granulocytic anaplasmosis was first reported in Anhui Province in 2006. From 2010 to 2015, 286 cases of severe fever with thrombocytopenia syndrome (SFTS) were diagnosed in Jiangsu and Anhui provinces, with a fatality rate of 16.1% [20]. Li et al. (2021) predicted that most areas of Shanghai are highly suitable for ticks, and previous investigations have shown that the dominant ticks in Shanghai are H. flava and H. longicornis [21]. However, no relevant investigations and research have been undertaken in recent decades. Therefore, we expect that the metagenomic analysis of Haemaphysalis spp. in this region may provide an extensive list of pathogens carried by this important vector, thereby highlighting the potential risk of human infection caused by tick bites.
In our study, we first investigate the bacterial variability between populations of H. flava and H. longicornis from Shanghai, China. We sequenced the amplicons of the eubacterial 16S rRNA to (1) determine the baseline bacterial diversity from ticks collected from within a relatively small geographic area, (2) confirm the species identity of key taxa using taxon-specific PCR and Sanger sequencing and (3) estimate the relative abundance of the key bacterial taxa by PCR in the pooled DNA of ticks collected from Shanghai, China.
2. Materials and Methods
2.1. Study Area
These ticks were collected from 18 sampling sites in Shanghai. Shanghai, China, is located in the front of the alluvial plain of the Yangtze River Delta, with soft soil and low and flat topography, with an average elevation of 4 m from east to west. Except for a few hills nearly 100 m above sea level in the west, Shanghai is a large and low-level plain, and the whole plain river port is similar to a net. Shanghai, China, belongs to the northern subtropical humid monsoon climate zone, warm, humid and rainy, with four distinct seasons. A suitable natural environment provides favorable conditions for the survival of ticks. The sample points are selected from random sites in each functional area of the city (Figure 1). (Central cities (CC): 121°45′ E, 31°16′ N; 121°37′ E, 31°19′ N; 121°47′ E, 31°22′ N; 121°45′ E, 31°28′ N; 121°41′ E, 31°23′ N; 121°48′ E, 31°24′ N; Out suburban districts (OSD): 121°21′ E, 31°11′ N; 121°19′ E, 31°08′ N; 121°48′ E, 30°92′ N; 121°18′ E, 31°07′ N; Inner suburban districts (ISD): 121°33′ E, 31°14′ N; 121°21′ E, 31°37′ N; 121°52′ E, 31°41′ N; 121°38′ E, 31°15′ N and Chongming island (CMI): 121°49′ E, 31°71′ N; 121°51′ E, 31°72′ N; 121°47′ E, 31°69′ N; 121°52′ E, 31°73′ N).
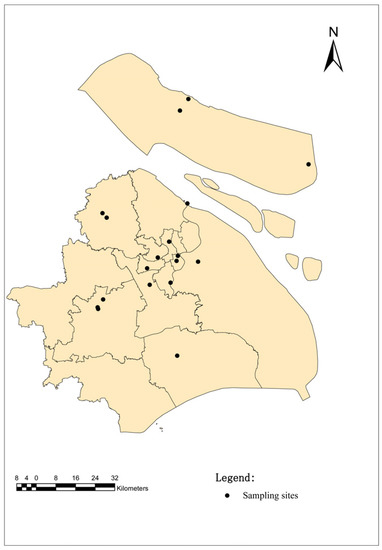
Figure 1.
Map of the sampling sites in Shanghai, China. The black dots indicate the sampling regions in this study.
2.2. Ticks Collection and Species Identification
Wild ticks were collected by dragging flags on the vegetation layer during the day. Additionally, parasitic ticks (90 H. flava and 22 H. longicornis) were collected from animals (dogs, goats, sheep, cows, etc.). For the first step, different morphological characteristics were observed to identify the species and development stages of the collected ticks by an entomologist (Zhu Dan) [22], and then 12S rDNA [23] and Cytochrome C oxidase subunit I (CO I) gene [24] identification was used to further determine the species of ticks, as previously described. All ticks of the genus Haemaphysalis from each site were included in the study. Secondly, in the lab stage, the ticks were rinsed with 75% ethanol for 1 min to remove any environmental contaminants; then, they were rinsed with deionized water for 5 min to remove 75% ethanol and finally stored in a refrigerator (−80 °C).
2.3. DNA Extraction
After morphological identification, the ticks collected from the same site were pooled according to developmental stages. Overall, 2102 H. flava (80 adults, 295 nymphs and 1727 larvae) were divided into 211 pools, and 151 H. longicornis (65 adults, 8 nymphs and 78 larvae) were divided into 15 pools. The total genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The DNA concentration and integrity were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis, respectively.
2.4. Molecular Identification of Tick Vectors by PCR
To further confirm the results of the morphological classification of Haemaphysalis spp., multi-locus sequence typing depending on five tick’s genomic DNA markers amplified fragments was carried out, which included: one nuclear gene CO I gene and a mitochondrial gene 12S rRNA. The PCR primers for the two genes are presented in Table 1.

Table 1.
Primers used in this study.
Table 1.
Primers used in this study.
| Target | Gene | Primer Name | Sequence (5′-3′) | Reference |
|---|---|---|---|---|
| Tick | 12S rDNA | T1B | AAACTAGGATTAGATACCCT | [23] |
| T2A | AATGAGAGCGACGGGCGATGT | |||
| Tick | CO I | HCO2198 | TAAACTTCAGGGTGACCAAAAAATCA | [24] |
| LCO1490 | GGTCAACAAATCATAAAGATATTGG | |||
| Microbiome | 16S rDNA | 343F | TACGGRAGGCAGCAG | [25] |
| 798R | AGGGTATCTAATCCT | |||
| Coxiella spp. | 16S rDNA | Cox16SF1 | CGTAGGAATCTACCTTRTAGWGG | [16] |
| Cox16SR1 | ACTYYCCAACAGCTAGTTCTCA | |||
| Cox16SF2 | TGAGAACTAGCTGTTGGRRAGT | |||
| Coc16SR2 | GCCTACCCGCTTCTGGTACAATT | |||
| Rickettsia spp. | ompA | Rr190.70p | ATGGCGAATATTTCTCCAAAA | [26] |
| Rr190.602n | AGTGCAGCATTCGCTCCCCCT | |||
| Ehrlichia spp. | 16S rRNA | Eh-out1 | TTGAGAGTTTGATCCTGGCTCAGAACG | [16,27] |
| Eh-out2 | CACCTCTACACTAGGAATTCCGCTATC | |||
| Eh-gs1 | GTAATACTGTATAATCCCTG | |||
| Eh-gs2 | GTACCGTCATTATCTTCCCTA | |||
| Anaplasma spp. | 16S rRNA | Eh-out1 | TTGAGAGTTTGATCCTGGCTCAGAACG | [27] |
| Eh-out2 | CACCTCTACACTAGGAATTCCGCTATC | |||
| HGA1 | GTCGAACGGATTATTCTTTATAGCTTG | |||
| HGA2 | TATAGGTACCGTCATTATCTTCCCTAC |
2.5. DNA Amplification
PCR amplification of the V3–V4 hypervariable regions of the bacterial 16S rRNA gene was carried out in a 25 μL reaction system using universal primer pairs (343F and 789R) (Table 1). The reverse primer contained a sample barcode, and both primers were connected with an Illumina sequencing adapter (Illumina Inc., San Diego, CA, USA).
2.6. Library Construction and Sequencing
The Amplicon quality was visualized using gel electrophoresis. The PCR products were purified with Agencourt AMPure XP beads (Beckman Coulter Co., Breya, CA, USA) and quantified using a Qubit dsDNA assay kit. The concentrations were then adjusted for sequencing. The sequencing was performed on an Illumina NovaSeq6000 with two paired-end read cycles of 250 bases each (Illumina Inc.; OE Biotech Company, Shanghai, China).
2.7. Bioinformatics Analysis
Raw sequencing data were in the FASTQ format. Paired-end reads were then pre-processed using cutadapt software to detect and cut off the adapter. After trimming, the paired-end reads were filtered for low-quality sequences, denoised, merged and detected and cut off the chimera reads using DADA2 [28] with the default parameters of QIIME2 [29] (November 2020); last, the software outputs, the representative reads and the ASV abundance table was generated. The representative read of each ASV was selected using the QIIME 2 package. All of the representative reads were annotated and blasted against Silva database Version 138 using q2-feature-classifier with the default parameters. The microbial diversity in the tick samples was estimated using the alpha diversity that includes the observed species index and Shannon index. The Unifrac distance matrix performed by QIIME software was used for unweighted Unifrac Principal coordinates analysis (PCoA).
2.8. Specific PCR for Detection of Some Pathogens in Ticks
Based on the results of 16S rRNA gene amplicon sequencing, genus-/group-specific PCR was performed to confirm the presence of TBPs in individual ticks. PCR was performed using a PCR System 9700 (Applied Biosystems, GeneAmp®, Carlsbad, CA, USA). For PCR, 2 μL of each DNA sample (150–330 ng) was used as the template for the first round, and 1 μL of the primary PCR product was used as the template for the second round. For the first round, a negative control (ddwater) and an extraction control mentioned above were included in each PCR experiment. Tube strips with individual caps were used in the amplification steps to prevent cross-contamination, and all PCR amplifications were carried out using PrimeSTAR® HS (Premix) (TaKaRa, Beijing, China). All of the operations were carried out in a biological safety cabinet. The amplified products were then electrophoresed on a 1.5% agarose gel, and the positive amplicons were sent to TSINGKE Biological Technology (Beijing, China) for sequencing. The PCR primers for Rickettsia spp., Anaplasma spp. and Ehrlichia spp. and Coxiella spp. are presented in Table 1.
2.9. Phylogenetic Analysis
The obtained nucleotide sequences were compared with those available in GenBank using the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) Basic Local Alignment Search Tool (BLAST) search engine (http://blast.ncbi.nlm.nih.gov/blast.cgi, accessed on 30 October 2022), and multiple sequence alignment was performed using the MEGA X (version 10.0) multiple alignment tool with the default parameters in MEGA X. The phylogenetic analysis was performed using MEGA X, and the tree was constructed using neighbor-joining (NJ) methods. The phylogenetic analysis of 12S rRNA and CO I gene for ticks, ompA for Rickettsia spp., 16S rRNA for Anaplasma spp., 16S rRNA for Ehrlichia spp., and 16S rRNA for Coxiella spp. was performed using the neighbor-joining method (NJ method) based on MEGA X. Bootstrap values were estimated for 1000 replicates.
3. Results
3.1. Taxonomic Classification and Sequencing Data Statistics
A total of 2253 hard ticks were identified as H. flava (n = 2102) and H. longicornis (n = 151) based on morphological identifications and confirmed by species-specific PCR and sequencing assays. There were 20 groups of H. flava (CCF1-5, OSDF1-5, ISDF1-5 and CMIF1-5) and 10 groups of H. longicornis (CCL1-5 and CMIL1-5) analyzed by amplicon sequencing on an Illumina NovaSeq6000 platform. The raw read data of the sequencing machine were distributed between 78,084 and 81,941, and the clean tag data after quality control were distributed between 4449 and 67,697. The valid tags (the final data used for analysis) data of clean tags were distributed between 4395 and 64,359, and the Amplicon Sequence Variant (ASV) numbers of each sample were distributed between 16 and 1191.
3.2. Alpha Diversity
Rarefaction curves were obtained for each tick group to determine if the sequencing depth was sufficient for each sample. Although the rarefaction curves for the observed ASVs approached saturation (Figure 2A), the Shannon diversity index curves reached a stable value (Figure 2B). The Good’s coverage values for each group of samples ranged from 91.69% to 100%, indicating that the majority of the ASVs had been discovered. Altogether, these results suggest that the sequencing depth was sufficient to represent the majority of bacterial communities in these samples.
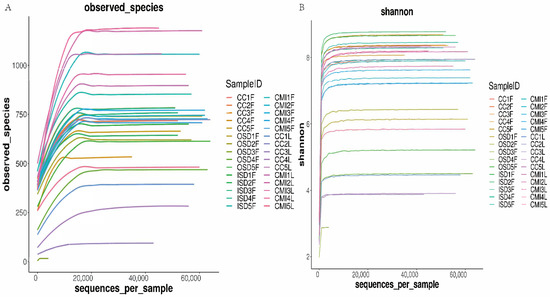
Figure 2.
Rarefaction curves of observed species (A) and Shannon diversity index (B) for group tick samples.
3.3. Bacterial Microbiome Composition
All of the valid sequences were classified with 100% identity, and species information was obtained by comparison with the SILVA-138SSUrRNA database. A total of 15,903 ASVs were detected in this study, with a total of 1872 species belonging to 33 phyla, 91 classes, 235 orders, 403 families and 946 genera. A total of 10674ASVs were detected in H. flava, with a total of 1872 species belonging to 32 phyla, 84 classes, 209 orders, 358 families and 801 genera. A total of 6010ASVs were detected in H. longicornis, with a total of 1162 species belonging to 31 phyla, 81 classes, 201 orders, 333 families and 696 genera.
Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria are the main components of bacterial communities of Haemaphysalis spp. The total species abundances of Proteobacteria, Firmicutes and Bacteroidetes, are 48.80%, 20.80% and 18.10%, respectively.
The bacteria in H. flava with high abundance include Proteobacteria, 48.80%; Firmicutes, 21.60%; Bacteroidetes, 17.70% and Actinobacteria, 6.70%. The bacteria in H. longicornis with high abundance include Proteobacteria, 48.8%; Firmicutes, 19.20%; Bacteroidetes, 19.00% and Actinobacteria, 9.00% (Figure 3).
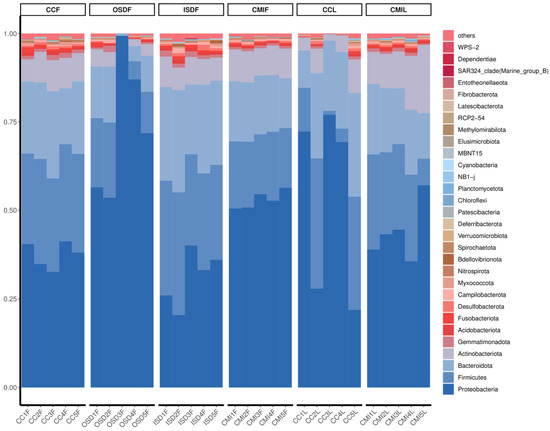
Figure 3.
Relative abundances of potential top 30 pathogens at the phylum level in grouped H. flava and H. longicornis samples. The groups of H. flava are CCF, OSDF, ISDF and CMIF, and the groups of H. longicornis are CCL and CMIL.
At the level of genus analysis, H. longicornis and H. flava carried at least 946 genera of bacteria. The bacteria in H. flava with high abundance include Lactobacillus, Rickettsia, Coxiella, Serratia and Muribaculaceae. The bacteria in H. longicornis with high abundance include Ochrobactrum, Coxiella, Lactobacillus and Sphingobacterium. Additionally, some groups of OSDF, CCF and CCL include Serratia (Figure 4).
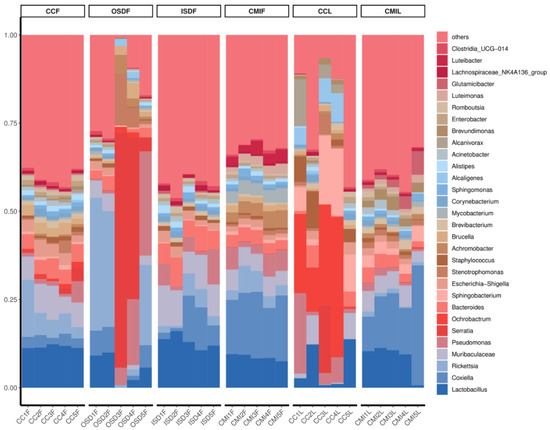
Figure 4.
Relative abundances of potential top 30 pathogens at genus level in grouped H. flava and H. longicornis samples. The groups of H. flava are CCF, OSDF, ISDF and CMIF and the groups of H. longicornis are CCL and CMIL.
3.4. Species and Location-Specific Differences in Microbial Diversity
We examined the effects of location and tick species on the bacterial diversity of the grouped samples. Diversity indices CCF, ISDF, CMIF and CMIL clustered tightly, while OSDF and CCL were more diffused (Variable), and some of them were significant (Figure 5). In the principal coordinates analysis, decreased variability could be accounted for across two axes (6.05% × 4.42%) (Figure 6). We detected a significant difference in the bacterial community between locations and species.
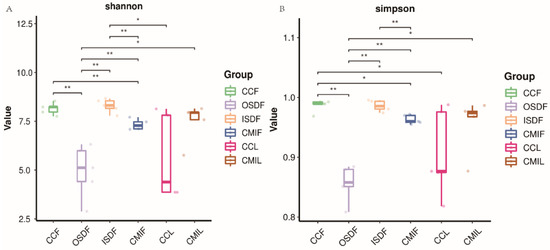
Figure 5.
Plots of Alpha diversity indices of grouped H. flava and H. longicornis samples. Plot (A) the Shannon diversity index and plots (B) the Simpson diversity index (* p < 0.05, ** p < 0.01). The groups of H. flava are CCF, OSDF, ISDF and CMIF and the groups of H. longicornis are CCL and CMIL.
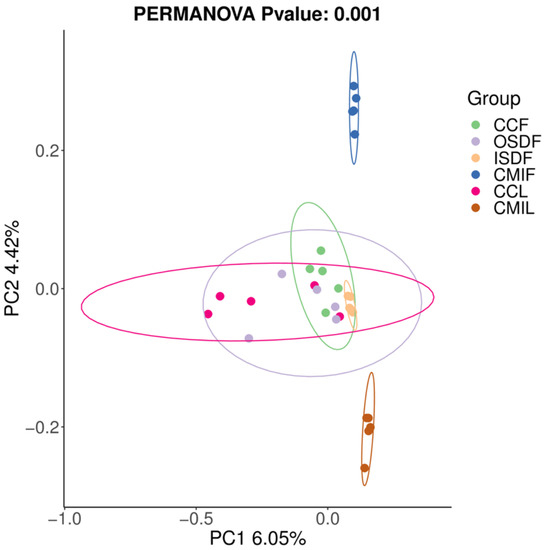
Figure 6.
Principal coordinates analysis (PCoA) plot of 16SrRNA data from grouped samples of H. flava and H. longicornis samples of sample-based ecological distance. The groups of H. flava are CCF, OSDF, ISDF and CMIF and the groups of H. longicornis are CCL and CMIL.
3.5. Bacterial Relative Abundance Differences
Of special interest is the identification of members of the genera Coxiella, Legionella, Anaplasma, Ehrlichia, Rickettsia and Sphingomonas, some of which are pathogenic and can be transmitted by ticks. The relative abundances of Lactobacillus, Coxiella, Sphingobacterium and Rickettsia differed significantly between the two groups (Figure 7).
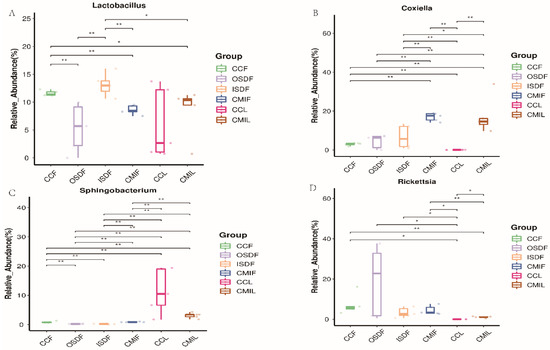
Figure 7.
Relative titers of bacterial 16SrRNA gene from Lactobacillus (A), Coxiella (B), Sphingobacterium (C) and Rickettsia (D) between groups of H. flava and H. longicornis samples (* p < 0.05, ** p < 0.01). The groups of H. flava are CCF, OSDF, ISDF and CMIF and the groups of H. longicornis are CCL and CMIL.
3.6. Prevalence of Tick-Borne Pathogens in Individual Pools
The important pathogenic bacterial genera Rickettsia and Coxiella were found in the grouped tick samples. Each pool was detected by the genus-/species-specific PCR combined with sequencing in order to identify the TBPs carried by it. In addition, Anaplasma and Ehrlichia were often detected in ticks, so each pool was screened by Anaplasma/Ehrlichia-specific PCR.
As a result, R. rickettsii (2.37%, 5/211), R. japonica (3.32%, 7/211), Candidatus R. jingxinensis (16.59%, 35/211), A. bovis (1.42%, 3/211), E. ewingii (0.95%, 2/211), E. chaffeensis (1.90%, 4/211), Coxiella spp. (1.90%, 4/211), C.-like endosymbiont (2.37%, 5/211) were detected in H. flava. from Shanghai and R. rickettsii (20.00%, 3/15), R. japonica (20.00%, 3/15), Candidatus R. jingxinensis (20.00%, 3/15), A. bovis (26.67%, 4/15), E. ewingii (13.33%, 2/15), E. chaffeensis (6.67%, 1/15), Coxiella spp. (6.67%, 1/15), C.-like endosymbiont (20.00%, 3/15) were also detected in H. longicornis from Shanghai (Table 2).
Table 2.
Prevalence of Rickettsia, Anaplasma, Ehrlichia and Coxiella in H. flava and H. longicornis.
3.7. Symbiotic Interaction of Bacterial Communities
The study also predicted the correlation among the top 30 genera in abundance. There is a positive correlation between Acinetobacter, Clostridia_UGG-014, Romboutsia, Corynebacterium, Enterobacter, Escherichia-Shigella, Lactobacillus, Bacteroides, Muribaculaceae, Alistipes and Lachnospiraceae_NK4A136_group. There is a negative correlation between one of Alcaligenes, Orchrobactrum, Achromobacter, Serratia, Stenotrophomonas and one of the above that has a positive correlation between them. There also is a positive correlation between Achromobacter, Serratia, Stenotrophomonas and Brucella, and there is a positive correlation between Coxilla, Luteimonas, Luteibacter and Mycobacterium (Figure 8).
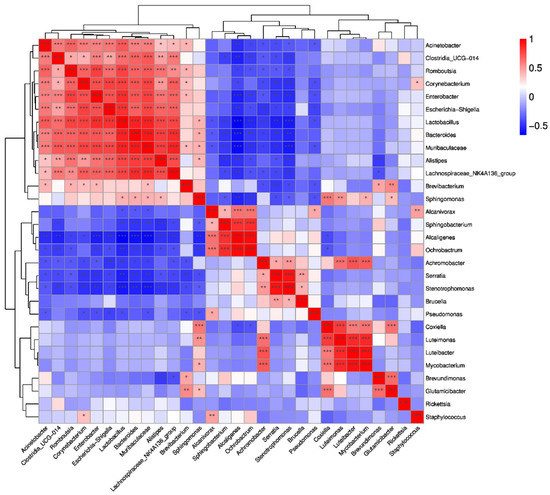
Figure 8.
Correlation heatmap of Spearman coefficient of the top 30 genera of the groups of H. flava and H. longicornis samples (* p < 0.05, ** p < 0.01,*** p < 0.001). The groups of H. flava are CCF, OSDF, ISDF and CMIF and the groups of H. longicornis are CCL and CMIL.
3.8. Phylogenetic Analysis
The phylogenetical analyses of 12S rDNA and CO I of the two tick species sequences (H. flava and H. longicornis) agreed with their morphological identification. Constructing a phylogenetic tree based on 12S rDNA, H. flava and H. longicornis were placed in the same clades with H. flava (OK054521, ON954856, KJ747360, JQ625665, MT013252, OM368276) and H. longicornis (JQ346678, KF583588, OM368281), respectively (Figure 9), and H. flava and H. longicornis were also placed in a clade with H. flava (MN066331, JQ737097, MN784164, MN650208) and H. longicornis (JQ737092), respectively, in the COI tree (Figure 10). By phylogenetic analysis, Coxiella spp. and C.-like endosymbiont, identified in both H. flava and H. longicornis, were shown to be clustered with Coxiella spp. (KC776319, MG906671) and C.-like endosymbiont (JQ480822), respectively (Figure 11). E. ewingii and E. chaffeensis, identified in both tick species in the study, were also placed in a clade with E. ewingii (NR_044747, U96436) and E. chaffeensis (AF147752, NR_074500, MZ433238) (Figure 12). A. bovis, identified in H. flava and H. longicornis, were shown to be clustered with A. bovis (GU556626) (Figure 13). R. japonica, R. rickettsii and Candidatus R. jingxinensis, identified in both tick species in the study, were also placed in a clade with R. japonica (U83440), R. rickettsii (AY319290) and Candidatus R. jingxinensis (MN550905) (Figure 14).
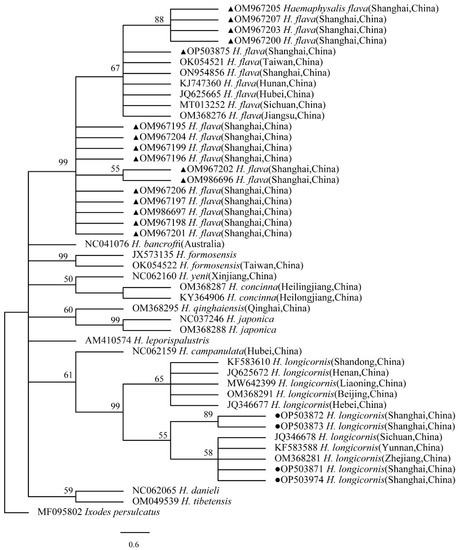
Figure 9.
Phylogenetic tree of H. flava and H. longicornis based on partial 12S rDNA gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. Kimura’s two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.
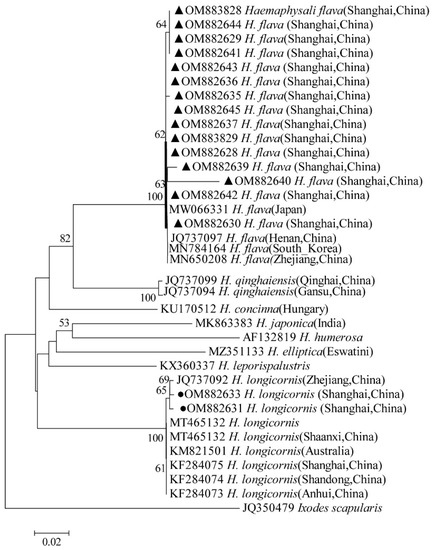
Figure 10.
Phylogenetic tree of H. flava and H. longicornis based on partial CO I gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. Kimura’s two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.
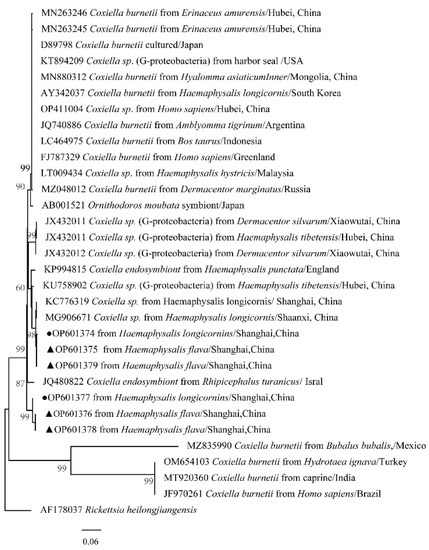
Figure 11.
Phylogenetic tree of Coxiella spp. in ticks based on partial 16S rDNA gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. The Kimura two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.
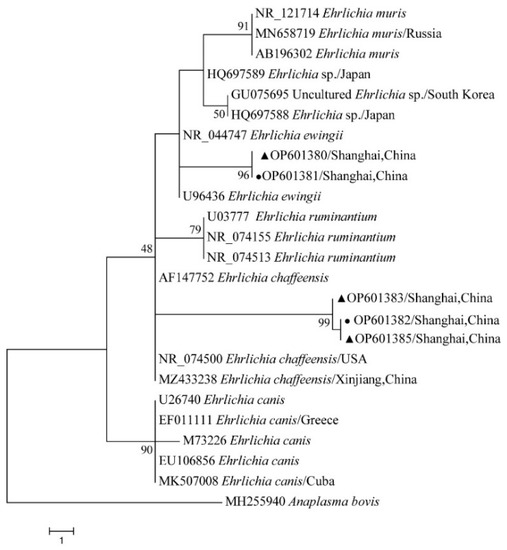
Figure 12.
Phylogenetic tree of Ehrlichia spp. in ticks based on partial 16S rDNA gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned by using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. The Kimura two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.

Figure 13.
Phylogenetic tree of Anaplasma spp. in ticks based on partial 16S rDNA gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. The Kimura two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.
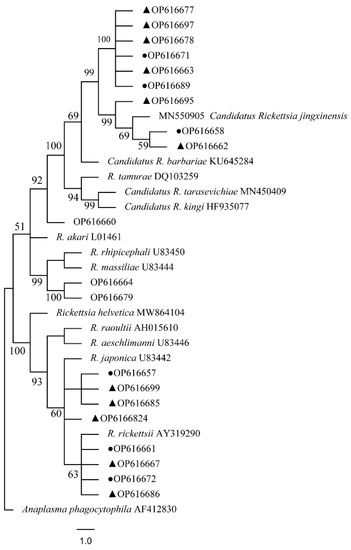
Figure 14.
Phylogenetic tree of Rickettsia spp. in ticks based on partial ompA gene sequence similarity. The sequence from H. flava obtained in this study is indicated with a black triangle, and the sequence from H. longicornis obtained in this study is indicated with black dots. Sequences were aligned using the MEGA X (version 10.0) software package. Phylogenetic analysis was performed by the neighbor-joining method (NJ method), and bootstrap values were estimated for 1000 replicates. The Kimura two-parameter model was used as a substitution model for the calculation of the phylogenetic trees.
4. Discussion
In recent years, more and more attention has focused on emerging TBPs and ticks. Additionally, a wide variety of pathogenic and non-pathogenic bacteria have been identified. In this study, the bacterial community diversity of Haemaphysalis spp. in Shanghai was analyzed and compared based on the 16S rDNA high-throughput sequencing technique combined with nested PCR to survey TBPs in Haemaphysalis spp. collected from Shanghai, China. It is found that the 16S rDNA V3–V4 variable region sequence of bacteria can effectively detect the bacterial community composition and diversity of H. flava and H. longicornis. At the same time, sequencing data can also be used to evaluate the relative abundance of bacteria and the differences in flora structure between different groups.
In this study, two species of Haemaphysalis ticks: H. longicornis and H. flava, were collected. H. flava often lives in mixed forests and fields, and their hosts are mainly pigs, pig badgers, horses and sheep, while H. longicornis mainly live in secondary forests, mountains, or hilly marginal areas, and their hosts are mainly cattle, horses, sheep, goats, bears, hedgehogs, etc.) [22]. However, in the past, H. longicornis was the main tick species in Shanghai [21], but today, H. flava, which has spread all over Shanghai, is the main tick species. In light of the ongoing geographical expansion of ticks caused by climatic change, as well as their increased ability to harbor new pathogens, public health concerns have been raised for both humans and animals [21,30,31,32]. Many new tick-borne pathogens have been discovered in recent decades, indicating the serious public health threat that tick-borne diseases have imposed on China [30]. The main pathogens reported in H. flava are Ehrlichia, Rickettsia japonica, Crimean–Congo hemorrhagic fever virus (CCHFV), Pseudomonas aeruginosa and Rickettsia raoultii [9,30]. Additionally, H. longicornis carries pathogens such as SFTSV, Anaplasma, spotted fever group Rickettsia (SFGR), Babesia, etc. [33,34]. The number of species of Haemaphysalis ticks in Shanghai is small, but its potential harm to human and animal husbandry should not be ignored.
In this study, the diversity of bacterial communities of H. flava and H. longicornis was different in species and regions. The α diversity analysis of metagenomics also showed that the combinations of tick bacteria were different with different biological factors (such as different developmental stages, age and sex) and differed from the tissue and environmental conditions investigated in other studies [17,35,36]. However, some regional differences were detected in most of the metagenomics β diversity indexes of male and female ticks [17], and any regional and gender differences were also found in the relative abundance of different bacterial groups [35,36,37,38]. The core bacterial communities may be influenced by maternal inheritance, the vertebrate animal skin microflora, host blood or the environment on physical contact [39,40]. However, a more specific and long-term association between some blood-feeding arthropods and their bacterial associate is likely mediated by the immune system of the host rather than by their external environment [40,41,42]. Bacterial taxa such as Bacillus, Clostridium, Methylobacterium, Mycobacterium, Pseudomonas, Sphingomonas and Staphylococcus, some of which were found in high abundance in our samples, contain species that may be commonly associated with the environment or as part of the mammalian skin flora [3,40,41,42]. Therefore, more stringent washing procedures, such as washing with sodium hypochlorite, or limiting sampling to internal organs, such as the salivary glands and mid-gut, may be necessary to determine the internal flora of ticks in further studies [3].
The bacteria in all the arthropod species were dominated by the phylum Proteobacteria, with proportions ranging from 48% to 72%, and their major bacteria phyla that were shared among all the arthropod species included Firmicutes, Bacteroidetes and Actinobacteria [40]. Our results are generally similar to those obtained in previous studies, where arthropod vectors species were dominated by Proteobacteria, including Gammaproteobacteria, Betaproteobacteria, Alphaproteobacteria, and to a lesser extent, Firmicutes, commonly Bacilli and Actinobacteria [40,43,44,45,46]. The taxonomic profiles at the genus level confirmed that genera Rickettsia, Anaplasma, Ehrlichia and Coxiella existed in the pooled sample, which all predominated in the top six in relative abundance [30]. The mutual connection of symbiotic bacteria hosts exists widely in nature. In recent years, the relationship between symbiotic bacteria and the host has received more and more attention; especially, the mode and mechanism of interaction between symbiotic bacteria and the host have aroused widespread concern. A past study has shown that Coxiella-like bacteria are frequently detected in Haemaphysalis ticks, although not all species carry them, and Coxiella-like endosymbionts have also been reported in other tick species such as H. shimoga, Rhipicephalus sanguineus, Amblyomma americanum, and the soft tick, Ornithodoros rostratus [3,47,48,49,50]. The significance of Coxiella in tick physiology is still unclear. Ticks may depend on bacteria, such as Coxiella-like bacteria and Coxiella-like endosymbionts (CLE), which play important roles in the processes of food digestion, biosynthesis, growth and development, reproductive regulation and the immune defense of ticks [51]. Studies have confirmed that the CLE genome has complete coding genes of the key coenzyme factor (B vitamins) biosynthesis pathway, which provides a stable source of vitamins for host ticks [52,53]. Removing CLE will significantly reduce the fertility of host ticks [53,54]. The host tick provides CLE with a stable growth environment and nutrition, which makes CLE grow and propagate in ticks and spread to offspring ticks. However, not all individuals of the same tick species will harbor endosymbionts at a similar abundance; some individuals may not carry any Coxiella-like endosymbionts at all according to other studies [3,54].
Coxiella-like bacteria, Legionella, Sphingomonas and other strains were detected. Coxiella-like bacteria are common in all tick samples, and the content is very high, which indicates that it may be an endosymbiont [55]. So far, in addition to focusing on the study of microbial communities and specific symbionts in ticks, attention has also been paid to bacterial interactions in ticks. There was a negative correlation between symbiotic bacteria and the serous edge of pathogens and a positive correlation among endosymbionts, Clostridium novyi and Corynebacterium cereus [56]. There also is a repulsion between symbiotic Rickettsia and pathogenic Rickettsia [35,57]. A detailed understanding of the interaction among microflora in ticks is helpful in developing new strategies for pathogens and tick vector control [17].
After sequencing the DNA fragments amplified by PCR and the sequence comparison, three Rickettsia species (Candidatus R. jingxinnensis, R. rickettsii and R. japonica), one Anaplasma species (A. bovis), two Coxiella species (Coxiella spp. and C.-like endosymbiont) and two Ehrlichia species (E. chaffeensis, E. ewingii) were found in both H. flava and H. longicornis. In Jiangsu province, China, which is close to Shanghai, Chian, R. japonica (81.1%), novel Rickettsia spp. (5.1%), A. bovis (12%), A. platys (6.3%), novel Ehrlichia spp. (16.6), C. burnetii (10.9%), and a novel Coxiella-like endosymbiont (CLE) strain (61.1%), detected in H. flava [30], are higher than in Shanghai, China. Especially, R. japonica has still been prevalent in China and Japan in recent years and can cause Oriental spotted fever. Interestingly, H. flava had a high positive rate (81.1%) of R. japonica in Jiangsu province, China [31], compared to the 3.32% detected in this tick species in Shanghai, China, in this study.
At present, there are three known species of Rickettsia japonica, Rickettsia rickettsii and Candidatus Rickettsia jingxinensis in Shanghai, as well as new species of Rickettsia that have not been identified or cultivated successfully, which have certain public health risks. Japanese spotted fever (JSF) caused by Rickettsia japonica infection is an acute febrile eruptive disease with ticks as the vector. The pathogen can also be detected in H. longicornis collected in Shandong, and researchers in Fujian province, China, have also amplified highly similar genes of Rickettsia japonica. Japan and South Korea have reported confirmed cases of Japanese spotted fever, and there are reports of Rickettsia japonica infection in Hebei, Anhui provinces, China [58]. Rocky Mountain Spotted Fever (RMSF) is caused by R. rickettsii infection. It has been observed that Candidatus R. jingxinensis can infect humans, showing the clinical features of fever, erythema rash and eschar [59]. Previous studies have shown that the main vectors of Rickettsia japonica are H. flava, H. formosensis, H. longicornis, H. cornigera, Ixodes ovatus, Rhipicephalus haemaphysaloides and Dermacentor taiwanensis. The vectors of RMSF are Amblyomma sculptum, Amblyomma aureolatum and Dermacento anderson. Candidatus R. jingxinensis were also detected in H. longicornis in Yunnan, Liaoning, Hebei and Jiangsu provinces, China, and Korea [60,61]. In Shanghai, Candidatus R. jingxinensis was detected in not only H. longicornis but also H. flava. In this study, Candidatus R. jingxinensis was detected from two tick species of Haemaphysalis spp., especially H. flava, which has spread all over Shanghai, China, which indicated that Candidatus R. jingxinensis had the characteristics of wide distribution. The deficiency of this study is that all three Rickettsia species have been detected in new tick species, but the genotype cannot be further confirmed and isolated by culture.
The two predominant human pathogens within the Ehrlichia genus are E. chaffeensis, the etiologic agent of human monocytic ehrlichiosis (HME) distributed in the United States, Asia and Europe, and E. ewingii, the agent of human E. ewingii granulocytic ehrlichiosis [62,63,64]. In southern and northern China, clues of E. chaffeensis were found, which indicated that E. chaffeensis might exist in China [65,66]. E. ewingii, one of the causative agents of canine granulocytic ehrlichiosis, has been reported in dogs and humans in the USA [67]. However, previous investigations and studies in China have found no clues of E. ewingii.
A. bovis is mainly distributed in Asia and South America. Cattle and buffalo are considered the main hosts of A. bovis, but A. bovis has also been found in dogs in China. The intangible disease is a tick-borne disease that is mainly prevalent in tropical and subtropical areas. It is not only healthy for livestock. Additionally, it will also threaten human health. Qin [41] investigated in Jiaonan, Shandong Province, China, the infection rate of the blood of ticks was found to be 0.10% for A. pagocytophilum, 1.55% for A. bovis and 0.33% for A. capra [68].
This study has shown that high-throughput 16S rDNA sequencing analysis can detect multiple pathogens at the same time, which can indicate the main local tick-borne pathogens, but it also has the disadvantage of incomplete detection and is unable to match species. Combined with pathogen-specific detection methods, we can more comprehensively understand the status of local Haemaphysalis spp.-carrying pathogens, so we need to combine PCR and metagenomics analysis.
In this study, it is worth noting that Serratia was detected in the medical tick group. Serratia infection can activate the mosquito’s immune system, significantly improve the mosquito’s resistance to Plasmodium berghei infection and reduce the malaria parasite load in Anopheles. A recent research report showed that Serratia isolated in the laboratory could promote insect-borne virus infection in mosquitoes [69,70]. This bacterium secretes a protein called SmEnhancin, which can bind mucin on intestinal mucosa to promote the colonization and spread of the virus in mosquitoes [71]. However, the research on ticks is blank, so we need to further study the interaction between ticks, hosts and the bacterial communities in ticks and pathogens and further explain their interaction mechanism.
5. Conclusions
This study reports on the bacterial communities and the prevalence of some TBPs in Haemaphysalis spp. from Shanghai, China. The results provide insight into the potential roles of Haemaphysalis ticks in the epidemiology of pathogens of veterinary and public health significance. Further studies are needed to elucidate the implications of these findings on animals and humans in Shanghai, China. However, these findings are preliminary. Further studies are required to determine the interaction and roles of some of the bacterial communities in tick survival and vector competence.
Author Contributions
Y.Z. and W.Z. designed the study. W.Z., Z.L., T.J., D.C., L.Y., T.H., L.D. and D.Z. collected the tick sample. W.Z. performed all the experiments and analyzed the data, and prepared draft figures. W.Z. and Z.L. prepared the manuscript draft with important intellectual input from Y.F. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financially sponsored by the Fifth Round of the Three-Year Action for Public Health System Construction in Shanghai (No. GWV-10.1-XK13); The Special Foundation of Basic Science and Technology Resources Survey of Ministry of Science and Technology of China (No. 2017FY101203); Shanghai sailing program (No. 21YF1452200).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The DNA sequencing data has been uploaded to GeneBank.
Acknowledgments
We are very grateful to the staff from Shanghai Center for Disease Control and Prevention, as well as Fan Mingqiu, Wen Heyi and Lin Chen for the tick sample collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dantas-Torres, F.; Chomel, B.B.; Otranto, D. Ticks and tick-borne diseases: A One Health perspective. Trends Parasitol. 2012, 28, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Mrzljak, A.; Novak, R.; Pandak, N.; Tabain, I.; Franusic, L.; Barbic, L.; Bogdanic, M.; Savic, V.; Mikulic, D.; Pavicic-Saric, J.; et al. Emerging and neglected zoonoses in transplant population. World J. Transplant. 2020, 10, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.J.; Chen, F.; Kho, K.L.; Shanizza, A.I.A.; Lim, F.S.; Tan, K.K.; Chang, L.Y.; AbuBakar, S. Bacterial community in Haemaphysalis ticks of domesticated animals from the Orang Asli communities in Malaysia. Ticks Tick Borne Dis. 2016, 7, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.R.; Fuerst, P.A. A rickettsial mixed infection in a Dermacentor variabilis tick from Ohio. Ann. N. Y. Acad. Sci. 2006, 1078, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.P.; Wang, Y.X.; Fan, Z.W.; Ji, Y.; Liu, M.J.; Zhang, W.H.; Li, X.L.; Zhou, S.X.; Li, H.; Liang, S.; et al. Mapping ticks and tick-borne pathogens in China. Nat. Commun. 2021, 12, 1075. [Google Scholar] [CrossRef]
- Wu, X.B.; Na, R.H.; Wei, S.S.; Zhu, J.S.; Peng, H.J. Distribution of tick-borne diseases in China. Parasites Vectors 2013, 6, 119. [Google Scholar] [CrossRef]
- Fang, L.Q.; Liu, K.; Li, X.L.; Liang, S.; Yang, Y.; Yao, H.W.; Sun, R.X.; Sun, Y.; Chen, W.J.; Zuo, S.Q.; et al. Emerging tick-borne infections in mainland China: An increasing public health threat. Lancet Infect. Dis. 2015, 15, 1467–1479. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.; Gao, X.; Bian, A.; Yan, H.; Kong, D.; Liu, X. Human Babesiosis in China: A systematic review. Parasitol. Res. 2019, 118, 1103–1112. [Google Scholar] [CrossRef]
- Fang, L.Z.; Lei, S.C.; Yan, Z.J.; Xiao, X.; Liu, J.W.; Gong, X.Q.; Yu, H.; Yu, X.J. Detection of Multiple Intracellular Bacterial Pathogens in Haemaphysalis flava Ticks Collected from Hedgehogs in Central China. Pathogens 2021, 10, 115. [Google Scholar] [CrossRef]
- Liu, L.M.; Liu, J.N.; Liu, Z.; Yu, Z.J.; Xu, S.Q.; Yang, X.H.; Li, T.; Li, S.S.; Guo, L.D.; Liu, J.Z. Microbial communities and symbionts in the hard tick Haemaphysalis longicornis (Acari: Ixodidae) from north China. Parasites Vectors 2013, 6, 310. [Google Scholar] [CrossRef]
- Tijsse-Klasen, E.; Hansford, K.M.; Jahfari, S.; Phipps, P.; Sprong, H.; Medlock, J.M. Spotted fever group rickettsiae in Dermacentor reticulatus and Haemaphysalis punctata ticks in the UK. Parasites Vectors 2013, 6, 212. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, H.; Wang, T.; Sun, W.; Yang, X.; Liu, J. Tick-borne pathogens and the vector potential of ticks in China. Parasites Vectors 2015, 8, 24. [Google Scholar] [CrossRef]
- Luo, J.; Ren, Q.; Liu, W.; Li, X.; Hong, Y.; Song, M.; Bo, Z.; Guan, G.; Luo, J.; Liu, G. Micropathogen community identification in ticks (Acari: Ixodidae) using third-generation sequencing. Int. J. Parasitol. Parasites Wildl. 2021, 15, 238–248. [Google Scholar] [CrossRef]
- Zhao, T.; Gong, H.; Shen, X.; Zhang, W.; Shan, T.; Yu, X.; Wang, S.J.; Cui, L. Comparison of Viromes in Ticks from Different Domestic Animals in China. Virol. Sin. 2020, 35, 398–406. [Google Scholar] [CrossRef]
- Ng, T.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS ONE 2011, 6, e20579. [Google Scholar] [CrossRef]
- Jiao, J.; Lu, Z.; Yu, Y.; Ou, Y.; Fu, M.; Zhao, Y.; Wu, N.; Zhao, M.; Liu, Y.; Sun, Y.; et al. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasites Vectors 2021, 14, 287. [Google Scholar] [CrossRef]
- Batool, M.; Blazier, J.C.; Rogovska, Y.V.; Wang, J.; Liu, S.; Nebogatkin, I.V.; Rogovskyy, A.S. Metagenomic analysis of individually analyzed ticks from Eastern Europe demonstrates regional and sex-dependent differences in the microbiota of Ixodes ricinus. Ticks Tick Borne Dis. 2021, 12, 101768. [Google Scholar] [CrossRef]
- Kodama, F.; Yamaguchi, H.; Park, E.; Tatemoto, K.; Sashika, M.; Nakao, R.; Terauchi, Y.; Mizuma, K.; Orba, Y.; Kariwa, H.; et al. A novel nairovirus associated with acute febrile illness in Hokkaido, Japan. Nat. Commun. 2021, 12, 5539. [Google Scholar] [CrossRef]
- Wang, Z.D.; Wang, B.; Wei, F.; Han, S.Z.; Zhang, L.; Yang, Z.T.; Yan, Y.; Lv, X.L.; Li, L.; Wang, S.C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Liu, Q.; He, B.; Huang, S.Y.; Wei, F.; Zhu, X.Q. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect. Dis. 2014, 14, 763–772. [Google Scholar] [CrossRef]
- Li, Z.Q.; Li, L.H.; Yin, H.J.; Wei, Z.X.; Guo, Y.H.; Ma, B.; Zhang, Y. Distribution and suitable habitats of ticks in the Yangtze River Delta urban agglomeration. Chin. J. Schistosomiasis Control 2021, 33, 365–372. [Google Scholar] [CrossRef]
- Yamaguti, N.; Tipton, V.J.; Keegan, H.L.; Toshioka, S. Ticks of Japan, Korea and the Ryukyu islands. Brighan Young Univ. Sci. Bull. 1971, 15, 1–226. [Google Scholar]
- Abdullah, H.H.; El-Molla, A.; Salib, F.A.; Allam, N.A.; Ghazy, A.A.; Abdel-Shafy, S. Morphological and molecular identification of the brown dog tick Rhipicephalus sanguineus and the camel tick Hyalomma dromedarii (Acari: Ixodidae) vectors of Rickettsioses in Egypt. Vet. World 2016, 9, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Chitimia, L.; Lin, R.Q.; Cosoroaba, I.; Wu, X.Y.; Song, H.Q.; Yuan, Z.G.; Zhu, X.Q. Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial cox1 and nad5 genes. Exp. Appl. Acarol. 2010, 52, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Nossa, C.W.; Oberdorf, W.E.; Yang, L.; Aas, J.A.; Paster, B.J.; Desantis, T.Z.; Brodie, E.L.; Malamud, D.; Poles, M.A.; Pei, Z. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 2010, 16, 4135–4144. [Google Scholar] [CrossRef]
- Regnery, R.L.; Spruill, C.L.; Plikaytis, B.D. Genotypic identification of rickettsiae and estimation of intraspecies sequence divergence for portions of two rickettsial genes. J. Bacteriol. 1991, 173, 1576–1589. [Google Scholar] [CrossRef]
- Cicuttin, G.L.; Brambati, D.F.; Rodríguez Eugui, J.I.; Lebrero, C.G.; De Salvo, M.N.; Beltrán, F.J.; Gury Dohmen, F.E.; Jado, I.; Anda, P. Molecular characterization of Rickettsia massiliae and Anaplasma platys infecting Rhipicephalus sanguineus ticks and domestic dogs, Buenos Aires (Argentina). Ticks Tick Borne Dis. 2014, 5, 484–488. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Qi, Y.; Ai, L.; Zhu, C.; Ye, F.; Lv, R.; Wang, J.; Mao, Y.; Lu, N.; Tan, W. Wild Hedgehogs and Their Parasitic Ticks Coinfected with Multiple Tick-Borne Pathogens in Jiangsu Province, Eastern China. Microbiol. Spectr. 2022, 10, e0213822. [Google Scholar] [CrossRef]
- Cun, D.J.; Wang, Q.; Yao, X.Y.; Ma, B.; Zhang, Y.; Li, L.H. Potential suitable habitats of Haemaphysalis longicornis in China under different climatic patterns. Chin. J. Schistosomiasis Control 2021, 33, 359–364. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Cui, X.; Jia, N.; Wei, J.; Xia, L.; Wang, H.; Zhou, Y.; Wang, Q.; Liu, X.; et al. Distribution of Haemaphysalis longicornis and associated pathogens: Analysis of pooled data from a China field survey and global published data. Lancet Planet. Health 2020, 4, e320–e329. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, K.; Cui, Y.; Zhou, Y.; Zhao, S.; Zhang, Y.; Jian, F.; Wang, R.; Zhang, L.; Ning, C. Molecular detection and phylogenetic analyses of Anaplasma spp. in Haemaphysalis longicornis from goats in four provinces of China. Sci. Rep. 2021, 11, 14155. [Google Scholar] [CrossRef]
- Jiang, J.; An, H.; Lee, J.S.; O’Guinn, M.L.; Kim, H.C.; Chong, S.T.; Zhang, Y.; Song, D.; Burrus, R.G.; Bao, Y.; et al. Molecular characterization of Haemaphysalis longicornis-borne rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 2018, 9, 1606–1613. [Google Scholar] [CrossRef]
- Gomard, Y.; Flores, O.; Vittecoq, M.; Blanchon, T.; Toty, C.; Duron, O.; Mavingui, P.; Tortosa, P.; McCoy, K.D. Changes in Bacterial Diversity, Composition and Interactions During the Development of the Seabird Tick Ornithodoros maritimus (Argasidae). Microb. Ecol. 2021, 81, 770–783. [Google Scholar] [CrossRef]
- Van Overbeek, L.; Gassner, F.; van der Plas, C.L.; Kastelein, P.; Nunes-da Rocha, U.; Takken, W. Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol. Ecol. 2008, 66, 72–84. [Google Scholar] [CrossRef]
- Carpi, G.; Cagnacci, F.; Wittekindt, N.E.; Zhao, F.; Qi, J.; Tomsho, L.P.; Drautz, D.I.; Rizzoli, A.; Schuster, S.C. Metagenomic profile of the bacterial communities associated with Ixodes ricinus ticks. PLoS ONE 2011, 6, e25604. [Google Scholar] [CrossRef]
- Thapa, S.; Zhang, Y.; Allen, M.S. Effects of temperature on bacterial microbiome composition in Ixodes scapularis ticks. MicrobiologyOpen 2019, 8, e00719. [Google Scholar] [CrossRef]
- Lim, F.S.; Khoo, J.J.; Tan, K.K.; Zainal, N.; Loong, S.K.; Khor, C.S.; AbuBakar, S. Bacterial communities in Haemaphysalis, Dermacentor and Amblyomma ticks collected from wild boar of an Orang Asli Community in Malaysia. Ticks Tick Borne Dis. 2020, 11, 101352. [Google Scholar] [CrossRef]
- Bennett, K.L.; Almanza, A.; McMillan, W.O.; Saltonstall, K.; Vdovenko, E.L.; Vinda, J.S.; Mejia, L.; Driesse, K.; De León, L.F.; Loaiza, J.R. Habitat disturbance and the organization of bacterial communities in Neotropical hematophagous arthropods. PLoS ONE 2019, 14, e0222145. [Google Scholar] [CrossRef]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The Tick Microbiome: Why Non-Pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Duguma, D.; Rugman-Jones, P.; Kaufman, M.G.; Hall, M.W.; Neufeld, J.D.; Stouthamer, R.; Walton, W.E. Bacterial communities associated with culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS ONE 2013, 8, e72522. [Google Scholar] [CrossRef] [PubMed]
- Menchaca, A.C.; Visi, D.K.; Strey, O.F.; Teel, P.D.; Kalinowski, K.; Allen, M.S.; Williamson, P.C. Preliminary assessment of microbiome changes following blood-feeding and survivorship in the Amblyomma americanum nymph-to-adult transition using semiconductor sequencing. PLoS ONE 2013, 8, e67129. [Google Scholar] [CrossRef]
- Muturi, E.J.; Ramirez, J.L.; Rooney, A.P.; Kim, C.H. Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl. Trop. Dis. 2017, 11, e0005377. [Google Scholar] [CrossRef]
- Osei-Poku, J.; Mbogo, C.M.; Palmer, W.J.; Jiggins, F.M. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 2012, 21, 5138–5150. [Google Scholar] [CrossRef]
- Ahantarig, A.; Malaisri, P.; Hirunkanokpun, S.; Sumrandee, C.; Trinachartvanit, W.; Baimai, V. Detection of Rickettsia and a novel Haemaphysalis shimoga symbiont bacterium in ticks in Thailand. Curr. Microbiol. 2011, 62, 1496–1502. [Google Scholar] [CrossRef]
- Noda, H.; Munderloh, U.G.; Kurtti, T.J. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl. Environ. Microbiol. 1997, 63, 3926–3932. [Google Scholar] [CrossRef]
- Jasinskas, A.; Zhong, J.; Barbour, A.G. Highly prevalent Coxiella sp. bacterium in the tick vector Amblyomma americanum. Appl. Environ. Microbiol. 2007, 73, 334–336. [Google Scholar] [CrossRef]
- Almeida, A.P.; Marcili, A.; Leite, R.C.; Nieri-Bastos, F.A.; Domingues, L.N.; Martins, J.R.; Labruna, M.B. Coxiella symbiont in the tick Ornithodoros rostratus (Acari: Argasidae). Ticks Tick Borne Dis. 2012, 3, 203–206. [Google Scholar] [CrossRef]
- Dall’Agnol, B.; McCulloch, J.A.; Mayer, F.Q.; Souza, U.; Webster, A.; Antunes, P.; Doyle, R.L.; Reck, J.; Ferreira, C.A.S. Molecular characterization of bacterial communities of two neotropical tick species (Amblyomma aureolatum and Ornithodoros brasiliensis) using rDNA 16S sequencing. Ticks Tick Borne Dis. 2021, 12, 101746. [Google Scholar] [CrossRef]
- Klyachko, O.; Stein, B.D.; Grindle, N.; Clay, K.; Fuqua, C. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl. Environ. Microbiol. 2007, 73, 6584–6594. [Google Scholar] [CrossRef]
- Smith, T.A.; Driscoll, T.; Gillespie, J.J.; Raghavan, R. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick. Genome Biol. Evol. 2015, 7, 831–838. [Google Scholar] [CrossRef]
- Tsementzi, D.; Castro Gordillo, J.; Mahagna, M.; Gottlieb, Y.; Konstantinidis, K.T. Comparison of closely related, uncultivated Coxiella tick endosymbiont population genomes reveals clues about the mechanisms of symbiosis. Environ. Microbiol. 2018, 20, 1751–1764. [Google Scholar] [CrossRef]
- Tufts, D.M.; Sameroff, S.; Tagliafierro, T.; Jain, K.; Oleynik, A.; VanAcker, M.C.; Diuk-Wasser, M.A.; Lipkin, W.I.; Tokarz, R. A metagenomic examination of the pathobiome of the invasive tick species, Haemaphysalis longicornis, collected from a New York City borough, USA. Ticks Tick Borne Dis. 2020, 11, 101516. [Google Scholar] [CrossRef]
- Gall, C.A.; Reif, K.E.; Scoles, G.A.; Mason, K.L.; Mousel, M.; Noh, S.M.; Brayton, K.A. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J. 2016, 10, 1846–1855. [Google Scholar] [CrossRef]
- Macaluso, K.R.; Sonenshine, D.E.; Ceraul, S.M.; Azad, A.F. Rickettsial infection in Dermacentor variabilis (Acari: Ixodidae) inhibits transovarial transmission of a second Rickettsia. J. Med. Entomol. 2002, 39, 809–813. [Google Scholar] [CrossRef]
- Li, W.; Liu, S.N. Rickettsia japonica infections in Huanggang, China, in 2021. IDCases 2021, 26, e01309. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.; Choi, Y.J.; Park, H.J.; Jang, W.J. Molecular genetic analysis and clinical characterization of Rickettsia species isolated from the Republic of Korea in 2017. Transbound. Emerg. Dis. 2020, 67, 1447–1452. [Google Scholar] [CrossRef]
- Qi, Y.; Ai, L.; Jiao, J.; Wang, J.; Wu, D.; Wang, P.; Zhang, G.; Qin, Y.; Hu, C.; Lv, R.; et al. High prevalence of Rickettsia spp. in ticks from wild hedgehogs rather than domestic bovine in Jiangsu province, Eastern China. Front. Cell. Infect. Microbiol. 2022, 12, 954785. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, J.; Choi, Y.J.; Kim, H.C.; Klein, T.A.; Chong, S.T.; Jiang, J.; Richards, A.L.; Jang, W.J. Tick-borne Rickettsiae in Midwestern region of Republic of Korea. Acta Trop. 2021, 215, 105794. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Yabsley, M.J.; Freye, J.D.; Dunlap, B.G.; Rowland, M.E.; Huang, J.; Dunn, J.R.; Jones, T.F.; Moncayo, A.C. Prevalence of Ehrlichia chaffeensis and Ehrlichia ewingii in ticks from Tennessee. Vector Borne Zoonotic Dis. 2010, 10, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Koh, F.X.; Kho, K.L.; Kisomi, M.G.; Wong, L.P.; Bulgiba, A.; Tan, P.E.; Lim, Y.A.L.; Nizam, Q.N.H.; Panchadcharam, C.; Tay, S.T. Ehrlichia and Anaplasma Infections: Serological Evidence and Tick Surveillance in Peninsular Malaysia. J. Med. Entomol. 2018, 55, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Kawahara, M.; Rikihisa, Y.; Fujita, H.; Watanabe, Y.; Suto, C.; Ito, T. New Ehrlichia species closely related to Ehrlichia chaffeensis isolated from Ixodes ovatus ticks in Japan. J. Clin. Microbiol. 2000, 38, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Zhang, L.X.; Li, W.H.; Wang, S.W.; Sun, Y.L.; Wang, Y.Y.; Guan, Z.Z.; Liu, X.J.; Yang, Y.S.; Zhang, S.G.; et al. Ehrlichiosis and zoonotic anaplasmosis in suburban areas of Beijing, China. Vector Borne Zoonotic Dis. 2012, 12, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.C.; Gao, Y.M.; Zhang, P.H.; Zhang, X.T.; Dai, Q.H.; Dumler, J.S.; Fang, L.Q.; Yang, H. Identification of Ehrlichia chaffeensis by nested PCR in ticks from Southern China. J. Clin. Microbiol. 2000, 38, 2778–2780. [Google Scholar] [CrossRef]
- Yabsley, M.J.; Varela, A.S.; Tate, C.M.; Dugan, V.G.; Stallknecht, D.E.; Little, S.E.; Davidson, W.R. Ehrlichia ewingii infection in white-tailed deer (Odocoileus virginianus). Emerg. Infect. Dis. 2002, 8, 668–671. [Google Scholar] [CrossRef]
- Qin, X.R.; Han, F.J.; Luo, L.M.; Zhao, F.M.; Han, H.J.; Zhang, Z.T.; Liu, J.W.; Xue, Z.F.; Liu, M.M.; Ma, D.Q.; et al. Anaplasma species detected in Haemaphysalis longicornis tick from China. Ticks Tick Borne Dis. 2018, 9, 840–843. [Google Scholar] [CrossRef]
- Bai, L.; Wang, L.; Vega-Rodríguez, J.; Wang, G.; Wang, S. A Gut Symbiotic Bacterium Serratia marcescens Renders Mosquito Resistance to Plasmodium Infection through Activation of Mosquito Immune Responses. Front. Microbiol. 2019, 10, 1580. [Google Scholar] [CrossRef]
- Bando, H.; Okado, K.; Guelbeogo, W.M.; Badolo, A.; Aonuma, H.; Nelson, B.; Fukumoto, S.; Xuan, X.; Sagnon, N.; Kanuka, H. Intra-specific diversity of Serratia marcescens in Anopheles mosquito midgut defines Plasmodium transmission capacity. Sci. Rep. 2013, 3, 1641. [Google Scholar] [CrossRef]
- Wu, P.; Sun, P.; Nie, K.; Zhu, Y.; Shi, M.; Xiao, C.; Liu, H.; Liu, Q.; Zhao, T.; Chen, X.; et al. A Gut Commensal Bacterium Promotes Mosquito Permissiveness to Arboviruses. Cell Host Microbe 2019, 25, 101–112.e105. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).