Recent Advances in Next Generation Snakebite Antivenoms



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
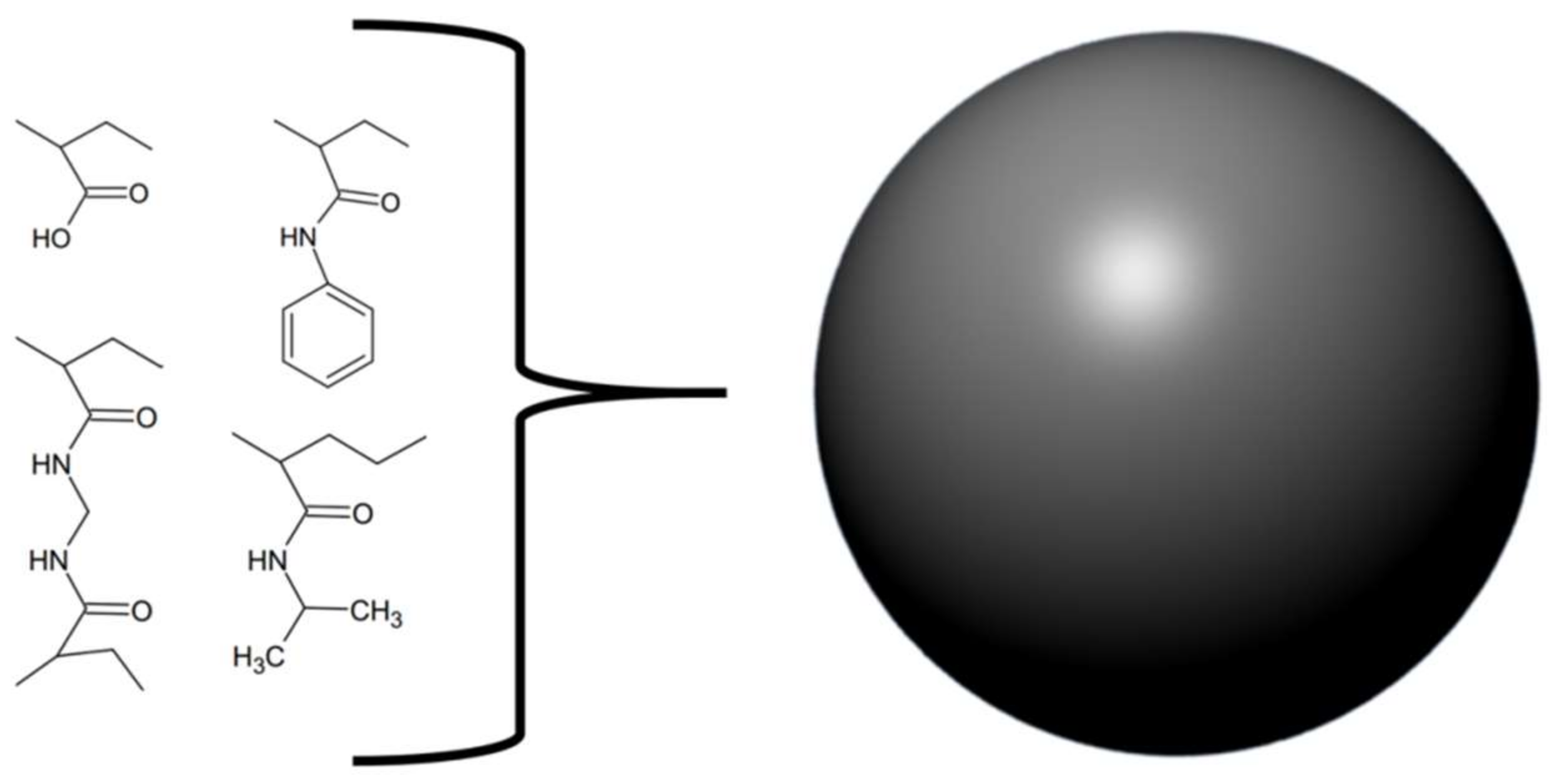
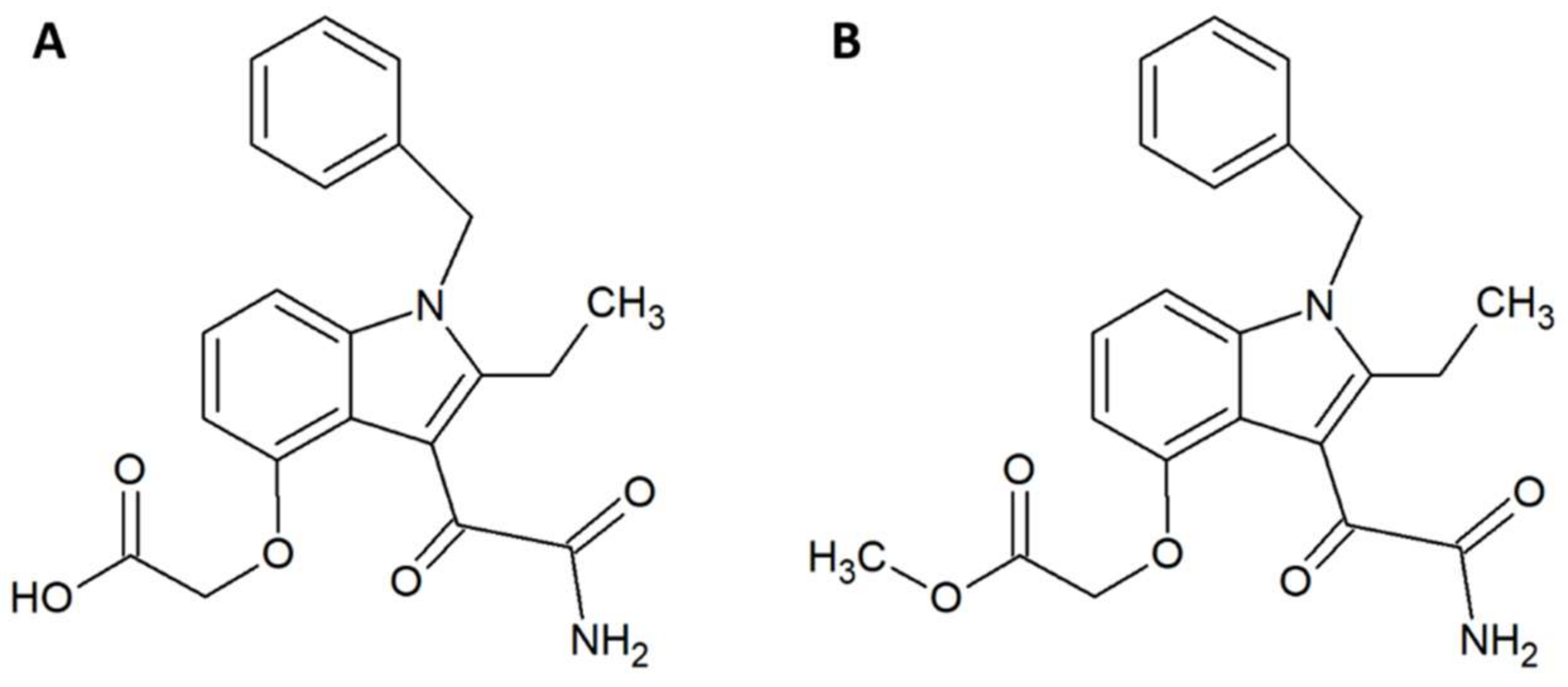
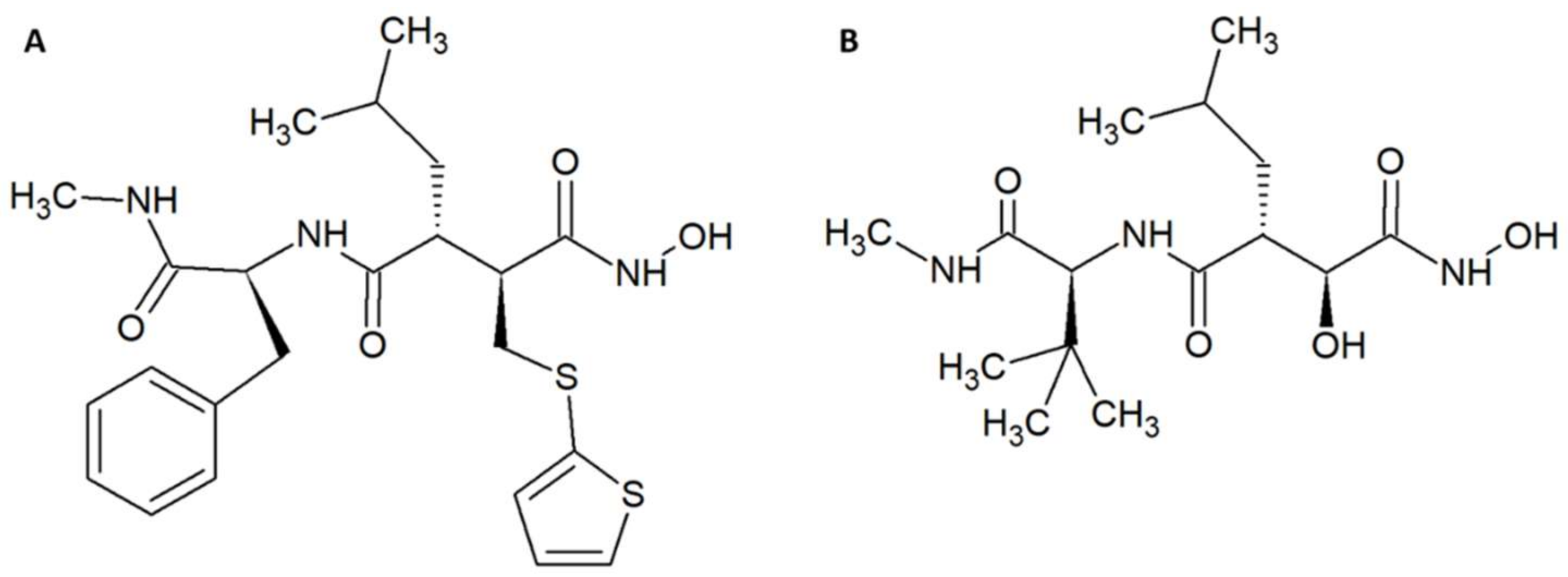
2. Small Molecule Inhibitors and Peptides
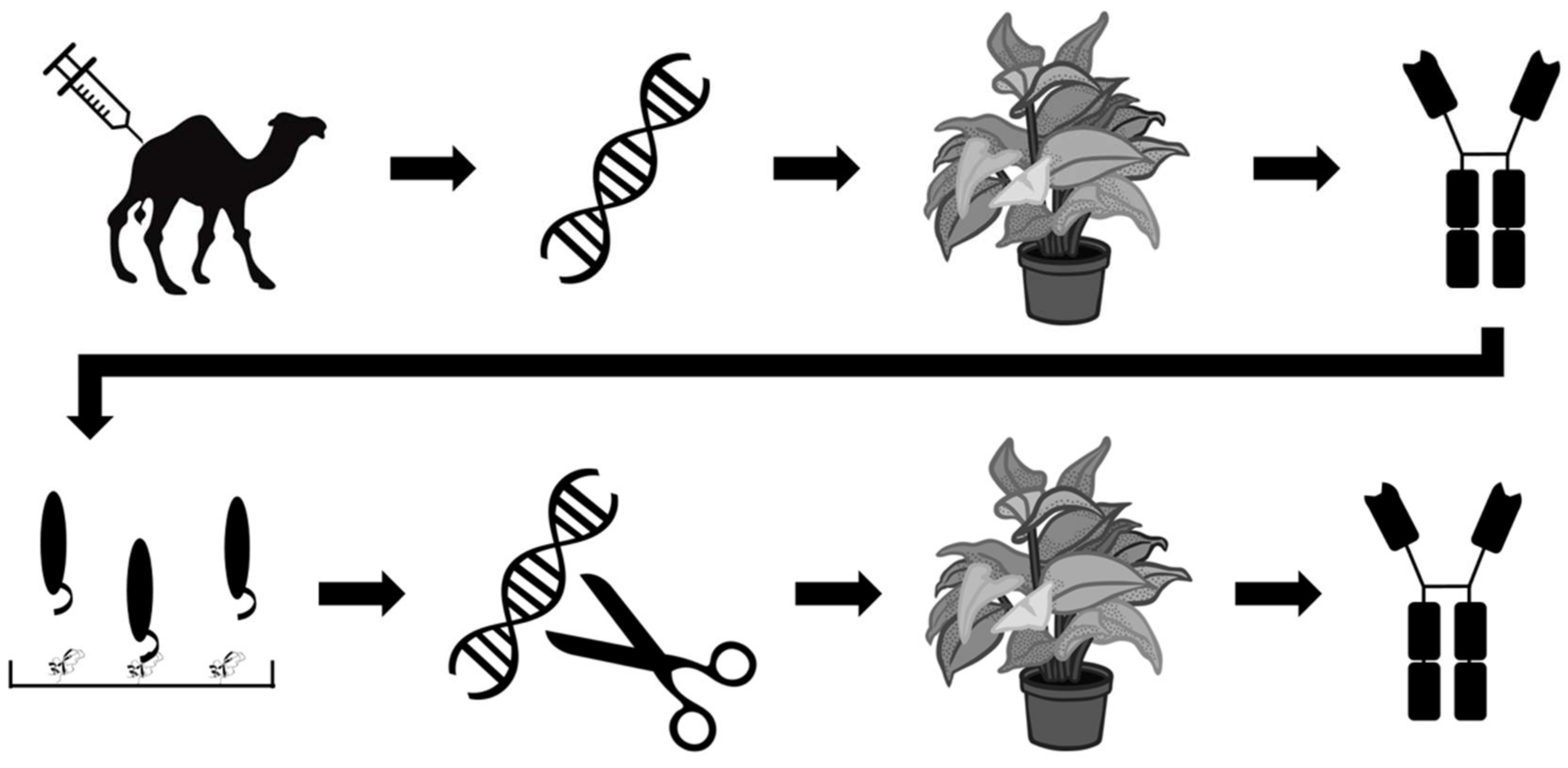
3. Oligonucleotides and Antibodies
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chippaux, J.-P. Snake-bites: Appraisal of the global situation. WHO Bull. OMS 1998, 76, 515–524. [Google Scholar]
- Kasturiratne, A.; Wickremasinghe, A.R.; De Silva, N.; Gunawardena, N.K.; Pathmeswaran, A.; Premaratna, R.; Savioli, L.; Lalloo, D.G.; De Silva, H.J. The global burden of snakebite: A literature analysis and modelling based on regional estimates of envenoming and deaths. PLoS Med. 2008, 5, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Chippaux, J.P. Estimate of the burden of snakebites in sub-Saharan Africa: A meta-analytic approach. Toxicon 2011, 57, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Mello, L.F.; Barcelos, M.G.; Meohas, W.; Pinto, L.W.; Melo, P.A.; Nogueira Neto, N.C.; Smith, J. Chronic ulceration of the leg following extensive scarring due to a snake bite complicated by squamous cell carcinoma. Skelet. Radiol. 2000, 29, 298–301. [Google Scholar] [CrossRef]
- Smith, J.; Mello, L.F.B.; Nogueira Neto, N.C.; Meohas, W.; Pinto, L.W.; Campos, V.A.; Barcellos, M.G.; Fiod, N.J.; Rezende, J.F.N.; Cabral, C.E.L. Malignancy in chronic ulcers and scars of the leg (Marjolin’s ulcer): A study of 21 patients. Skelet. Radiol. 2001, 30, 331–337. [Google Scholar] [CrossRef]
- Warrell, D.A.; Ormerod, L.D. Snake venom ophthalmia and blindness caused by the spitting cobra (Naja nigricollis) in Nigeria. Am. J. Trop. Med. Hyg. 1976, 25, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.S.; Wijesinghe, C.A.; Jayamanne, S.F.; Buckley, N.A.; Dawson, A.H.; Lalloo, D.G.; de Silva, H.J. Delayed psychological morbidity associated with snakebite envenoming. PLoS Negl. Trop. Dis. 2011, 5, e1255. [Google Scholar] [CrossRef] [PubMed]
- Theakston, R.; Warrell, D. Crisis in snake antivenom supply for Africa. Lancet 2000, 356, 2104. [Google Scholar] [CrossRef]
- Williams, D.J.; Gutiérrez, J.; Calvete, J.J.; Wüster, W.; Ratanabanangkoon, K.; Paiva, O.; Brown, N.I.; Casewell, N.R.; Harrison, R.A.; Rowley, P.D.; et al. Ending the drought: New strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteom. 2011, 74, 1735–1767. [Google Scholar] [CrossRef] [PubMed]
- Ariaratnam, C.A.; Sjostrom, L.; Raziek, Z.; Kularatne, S.A.; Arachchi, R.W.K. Kodikara Sheriff, M.H.R.; Theakston, R.D.G.; Warrell, D.A. An open, randomized comparative trial of two antivenoms for the treatment of envenoming by Sri Lankan Russell’ s viper (Daboia russelii). Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 74–80. [Google Scholar] [CrossRef]
- León, G.; Monge, M.; Rojas, E.; Lomonte, B.; Gutiérrez, J.M. Comparison between IgG and F(ab′)2 polyvalent antivenoms: Neutralization of systemic effects induced by Bothrops asper venom in mice, extravasation to muscle tissue, and potential for induction of adverse reactions. Toxicon 2001, 39, 793–801. [Google Scholar] [CrossRef]
- Herrera, M.; León, G.; Segura, A.; Meneses, F.; Lomonte, B.; Chippaux, J.P.; Gutiérrez, J.M. Factors associated with adverse reactions induced by caprylic acid-fractionated whole IgG preparations: Comparison between horse, sheep and camel IgGs. Toxicon 2005, 46, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.E.; Kyei-Faried, S.; Belcher, D.W.; Geelhoed, D.W.; van Leeuwen, J.S.; van Roosmalen, J. Failure of a new antivenom to treat Echis ocellatus snake bite in rural Ghana: The importance of quality surveillance. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Arias, A.S.; Rodríguez, Y.; Quesada-Bernat, S.; Sánchez, L.V.; Chippaux, J.P.; Pla, D.; Gutiérrez, J.M. Preclinical evaluation of three polyspecific antivenoms against the venom of Echis ocellatus: Neutralization of toxic activities and antivenomics. Toxicon 2016, 119, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Bochner, R. Paths to the discovery of antivenom serotherapy in France. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chippaux, J.P. WHO guidelines for the production, control and regulation of snake antivenom immunoglobulins. Biol. Aujourdhui 2010, 204, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Engmark, M.; Milbo, C.; Johannesen, J.; Lomonte, B.; Gutiérrez, J.M.; Lohse, B. From fangs to pharmacology: The future of antivenoms. Curr. Pharm. Des. 2016, 9, 5270–5293. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Solà, M.; Jappe, E.C.; Oscoz, S.; Lauridsen, L.P.; Engmark, M. Biotechnological trends in spider and scorpion antivenom development. Toxins (Basel) 2016, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Guiding recombinant antivenom development by omics technologies. N. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Gutiérrez, J.M.; Knudsen, C.; Johansen, K.H.; Méndez, E.B.; Cerni, F.A.; Jürgensen, J.A.; Øhlenschlæger, M.; Ledsgaard, L.; Esteban, A.M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018. [CrossRef] [PubMed]
- Gutiérrez, J.M.; León, G.; Lomonte, B.; Angulo, Y. Antivenoms for snakebite envenomings. Inflamm. Allergy Drug Targets 2011, 10, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Chippaux, J.P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Rägo, L.; Marroquin, A.M.P.; Nübling, C.M.; Sawyer, J. Treating snake bites—A call for partnership. Lancet 2015, 386, 2252. [Google Scholar] [CrossRef]
- Williams, D.J. Snake bite: A global failure to act costs thousands of lives each year. BMJ 2015, 351, h5378. [Google Scholar] [CrossRef] [PubMed]
- Alirol, E.; Lechevalier, P.; Zamatto, F.; Chappuis, F.; Alcoba, G.; Potet, J. Antivenoms for snakebite envenoming: What is in the research pipeline? PLoS Negl. Trop. Dis. 2015, 9, e0003896. [Google Scholar] [CrossRef] [PubMed]
- Word Health Organization WHO Working Group on Snakebite Envenoming. Available online: http://www.who.int/snakebites/control/WHO_Working_Group_on_Snakebite_Envenoming/en/ (accessed on 8 April 2018).
- U.S. National Library of Medicine Vista-16 trial: Evaluation of Safety and Efficacy of Short-term A-002 Treatment in Subjects With Acute Coronary Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT01130246 (accessed on 8 April 2018).
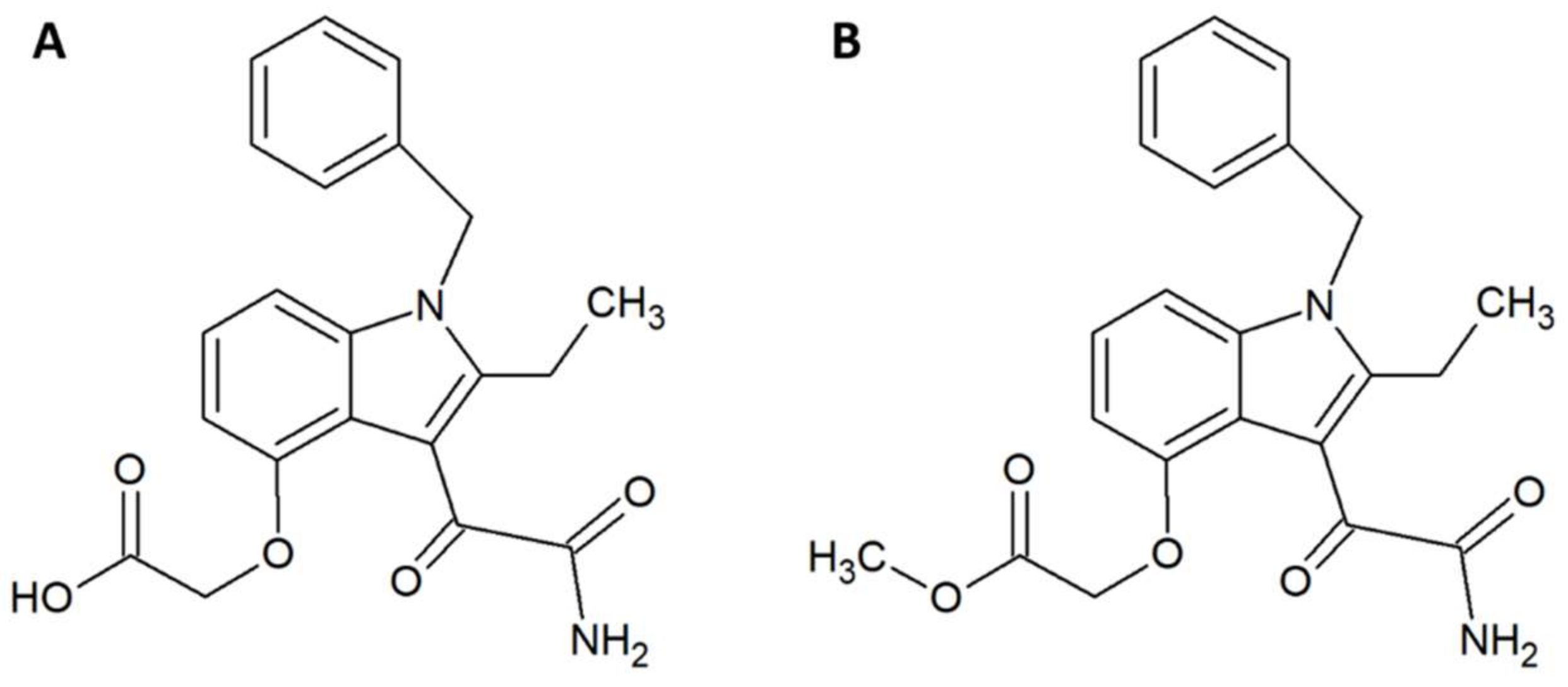
- Lewin, M.; Samuel, S.; Merkel, J.; Bickler, P. Varespladib (LY315920) appears to be a potent, broad-spectrum, inhibitor of snake venom phospholipase A2 and a possible pre-referral treatment for envenomation. Toxins (Basel) 2016, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Zhang, D.; Xiao, H.; Xiong, S.; Huang, C. exploration of the inhibitory potential of varespladib for snakebite envenomation. Molecules 2018, 23, 391. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Engmark, M.; Clouser, C.; Timberlake, S.; Vigneault, F.; Gutiérrez, J.M.; Lomonte, B. Exploration of immunoglobulin transcriptomes from mice immunized with three-finger toxins and phospholipases A2 from the Central American coral snake, Micrurus nigrocinctus. Peer J 2017, 5, e2924. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Toxin synergism in snake venoms. Toxin Rev. 2016, 35, 165–170. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Lomonte, B.; Lohse, B.; Fernández, J.; Gutiérrez, J.M. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: Identification of key toxin targets for antivenom development. J. Proteom. 2015, 119, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, L.P.; Laustsen, A.H.; Lomonte, B.; Gutiérrez, J.M. Toxicovenomics and antivenom profiling of the eastern green mamba snake (Dendroaspis angusticeps). J. Proteom. 2016, 136, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, S.; Petras, D.; Engmark, M.; Süssmuth, R.D.; Whiteley, G.; Albulescu, L.O.; Kazandjian, T.D.; Wagstaff, S.C.; Rowley, P.; Wüster, W.; et al. The medical threat of mamba envenoming in sub-Saharan Africa revealed by genus-wide analysis of venom composition, toxicity and antivenomics profiling of available antivenoms. J. Proteom. 2017, 172, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Escalante, T.; Franceschi, A.; Rucavado, A.; Gutiérrez, J.M. Effectiveness of batimastat, a synthetic inhibitor of matrix metalloproteinases, in neutralizing local tissue damage induced by BaP1, a hemorrhagic metalloproteinase from the venom of the snake Bothrops asper. Biochem. Pharmacol. 2000, 60, 269–274. [Google Scholar] [CrossRef]
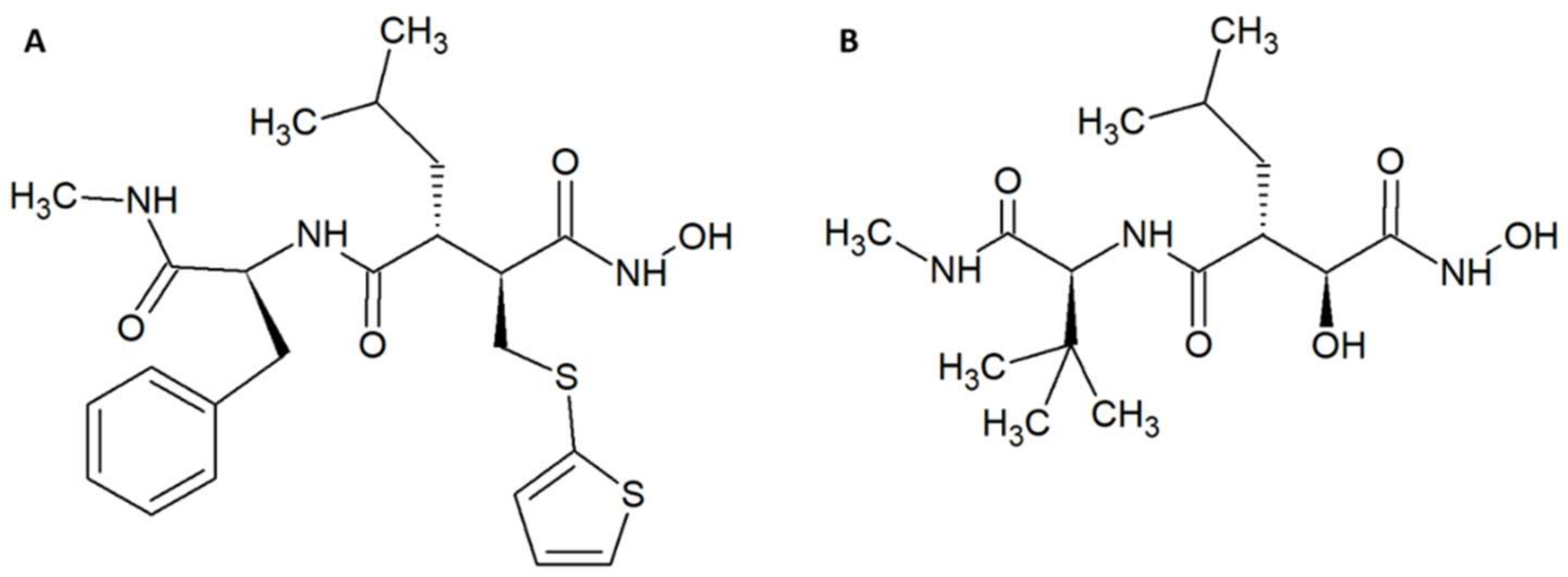
- Villalta-Romero, F.; Gortat, A.; Herrera, A.E.; Arguedas, R.; Quesada, J.; De Melo, R.L.; Calvete, J.J.; Montero, M.; Murillo, R.; Rucavado, A.; et al. Identification of new snake venom metalloproteinase inhibitors using compound screening and rational peptide design. ACS Med. Chem. Lett. 2012, 3, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.S.; Rucavado, A.; Gutiérrez, J.M. Peptidomimetic hydroxamate metalloproteinase inhibitors abrogate local and systemic toxicity induced by Echis ocellatus (saw-scaled) snake venom. Toxicon 2017, 132, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Flipo, M.; Charton, J.; Hocine, A.; Dassonneville, S.; Deprez, B.; Deprez-Poulain, R. Hydroxamates: Relationships between structure and plasma stability. J. Med. Chem. 2009, 52, 6790–6802. [Google Scholar] [CrossRef] [PubMed]
- Beattie, G.J.; Smyth, J.F. Phase I Study of intraperitoneal metalloproteinase inhibitor BB94 in patients with maglinant ascites. Clin. Cancer Res. 1998, 4, 1899–1902. [Google Scholar] [PubMed]
- Millar, A.W.; Brown, P.D.; Moore, J.; Galloway, W.A.; Cornish, A.G.; Lenehan, T.J.; Lynch, K.P. Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br. J. Clin. Pharmacol. 1998, 45, 21–26. [Google Scholar] [CrossRef] [PubMed]
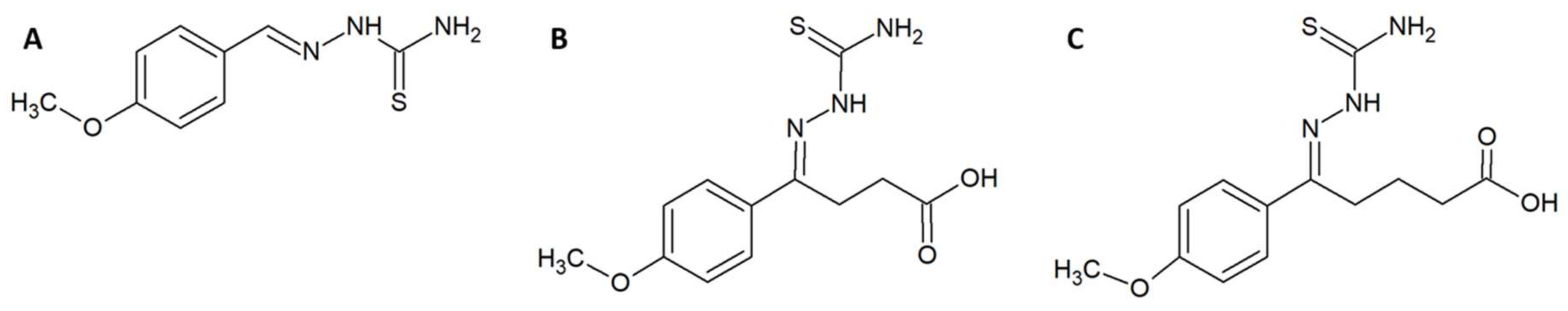
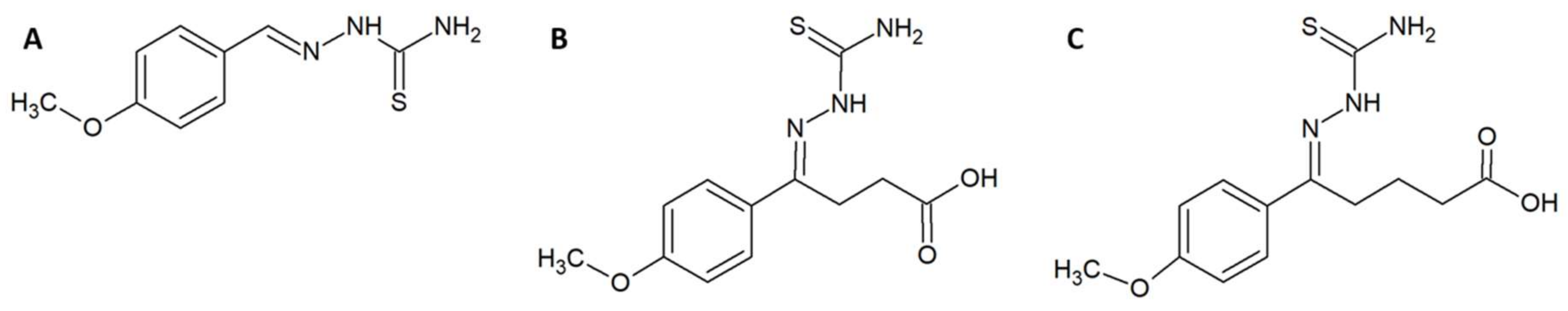
- Ferreira, F.B.; Pereira, T.M.; Souza, D.L.N.; Lopes, D.S.; Freitas, V.; Ávila, V.M.R.; Kümmerle, A.E.; Sant’Anna, C.M.R. Structure-based discovery of thiosemicarbazone metalloproteinase inhibitors for hemorrhage treatment in snakebites. ACS Med. Chem. Lett. 2017, 8, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Recombinant Antivenoms. Ph. D. thesis, University of Copenhagen, Copenhagen, Denmark, 2016. [Google Scholar]
- Titus, J.K.; Kay, M.K.; Glaser, C.D.R.J.J.; Hwang, Y.Y. Application of phage display for the development of a novel inhibitor of PLA 2 activity in western cottonmouth venom. J. Venom Res. 2017, 8, 19–24. [Google Scholar] [PubMed]
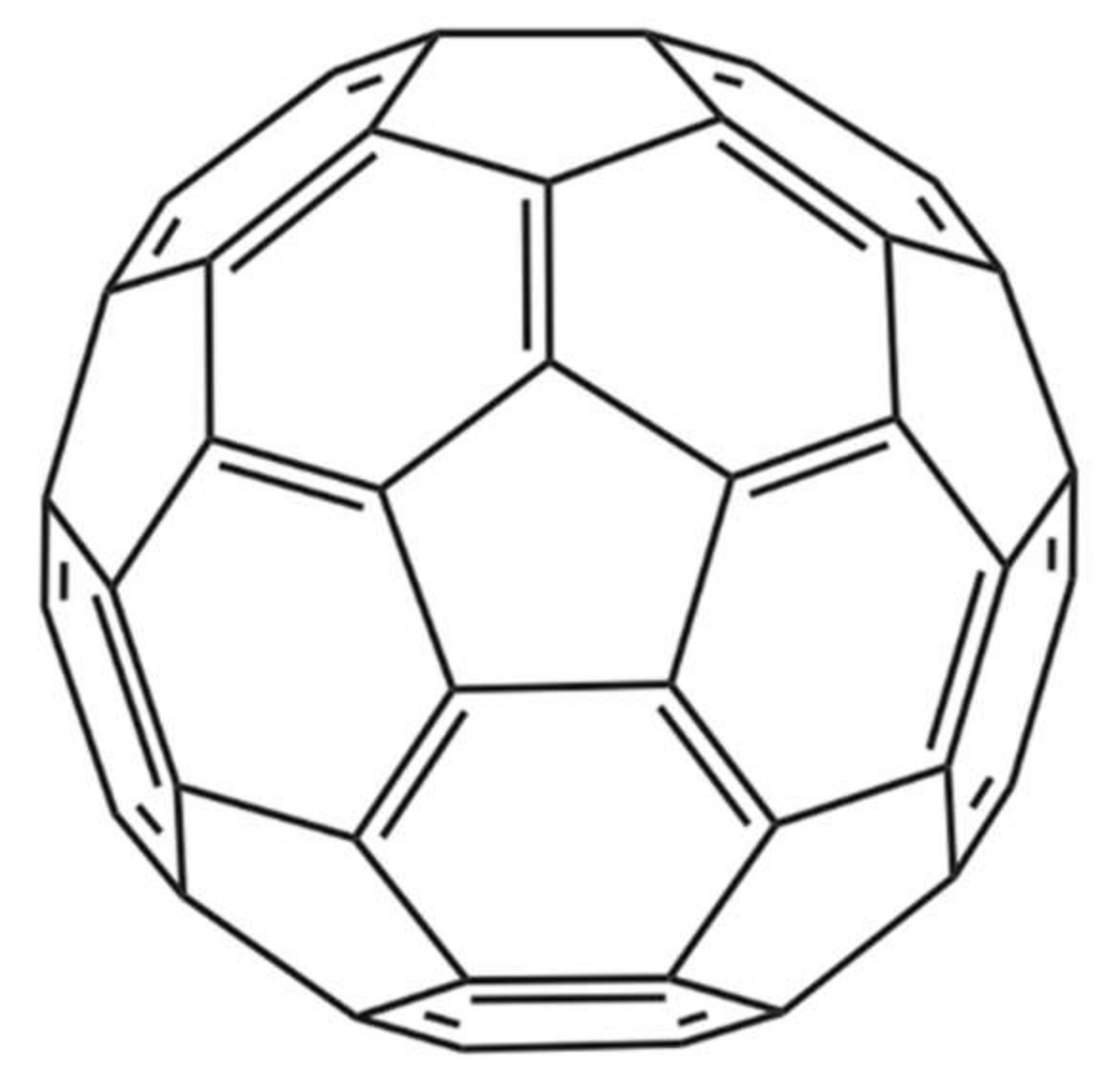
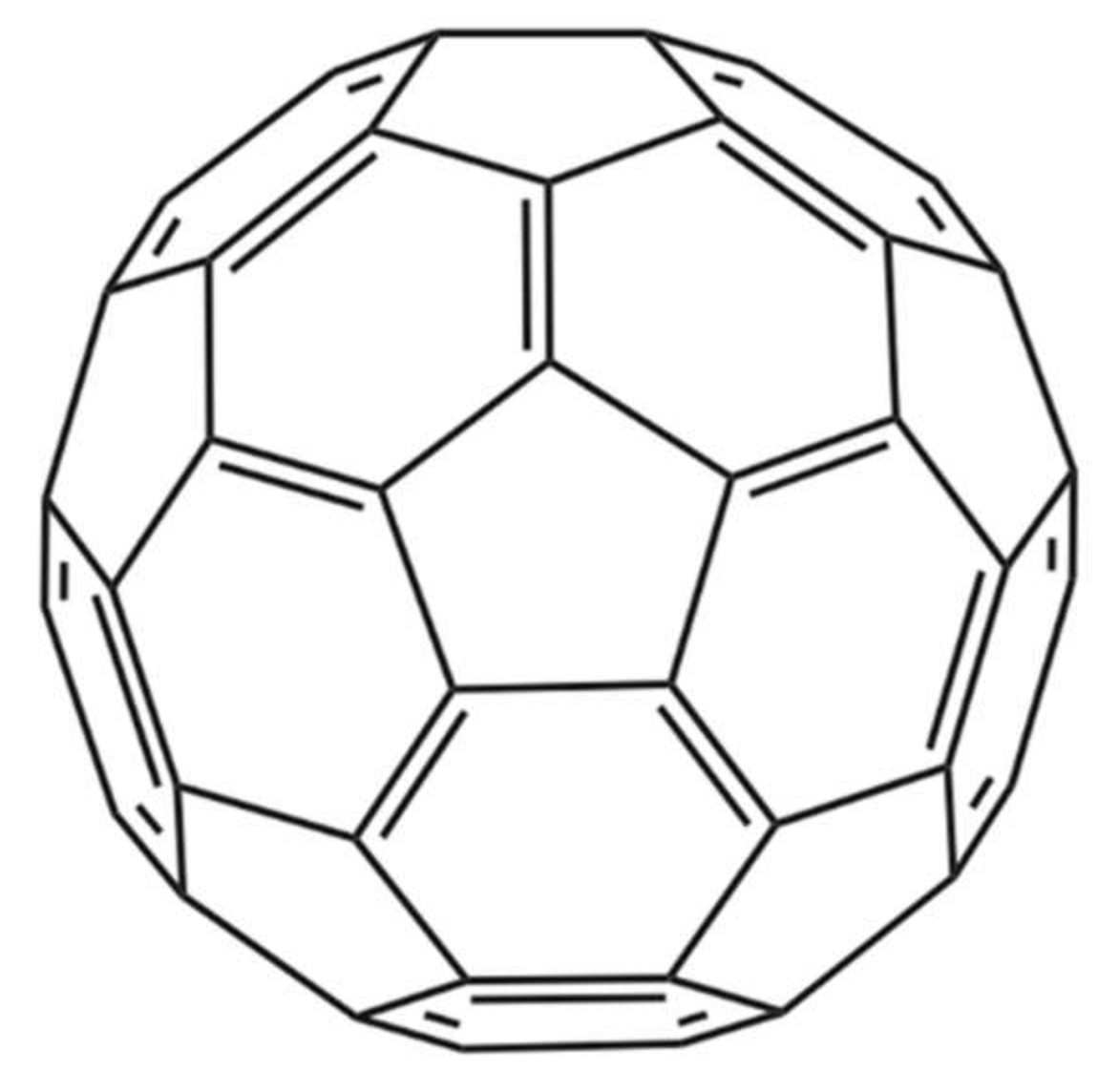
- Karain, B.D.; Lee, M.K.H.; Hayes, W.K. C60 Fullerenes as a novel treatment for poisoning and envenomation: A proof-of-concept study for snakebite. J. Nanosci. Nanotechnol. 2016, 16, 7764–7771. [Google Scholar] [CrossRef]
- Wikimedia Commons. Buckminsterfullerene. Available online: https://commons.wikimedia.org/wiki/File:Buckminsterfullerene.svg (accessed on 8 April 2018).
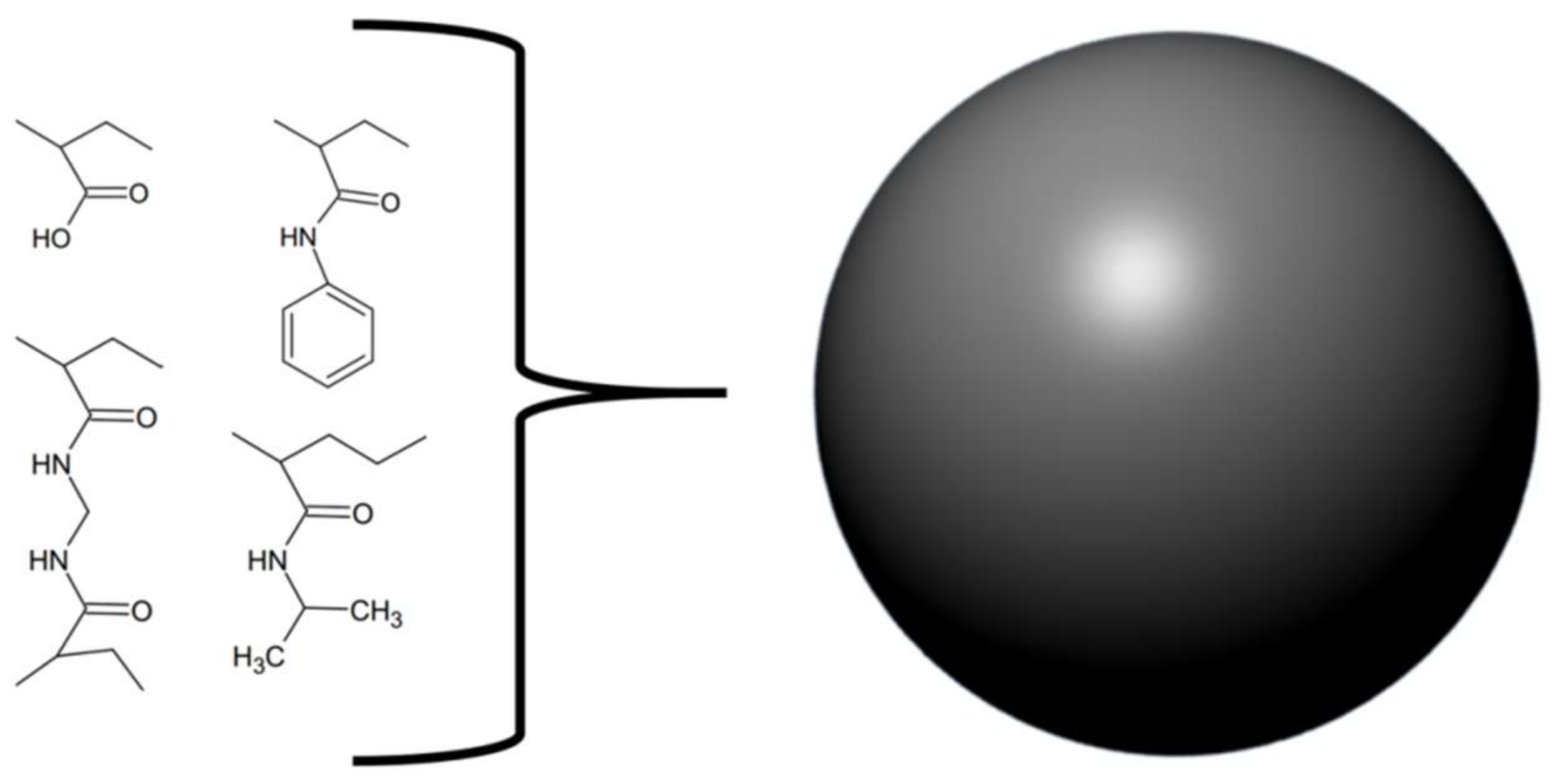
- O’Brien, J.; Lee, S.-H.; Onogi, S.; Shea, K.J. Engineering the protein corona of a synthetic polymer nanoparticle for broad-spectrum sequestration and neutralization of venomous biomacromolecules. J. Am. Chem. Soc. 2016, 138, 16604–16607. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.K.; Bruno, J.G.; Dhiman, A. ABCs of DNA aptamer and related assay development. Biotechnol. Adv. 2017, 35, 275–301. [Google Scholar] [CrossRef] [PubMed]
- El-Aziz, T.M.A.; Ravelet, C.; Molgo, J.; Fiore, E.; Pale, S.; Amar, M.; Al-Khoury, S.; Dejeu, J.; Fadl, M.; Ronjat, M.; et al. Efficient functional neutralization of lethal peptide toxins in vivo by oligonucleotides. Sci. Rep. 2017, 7, 7202. [Google Scholar] [CrossRef] [PubMed]
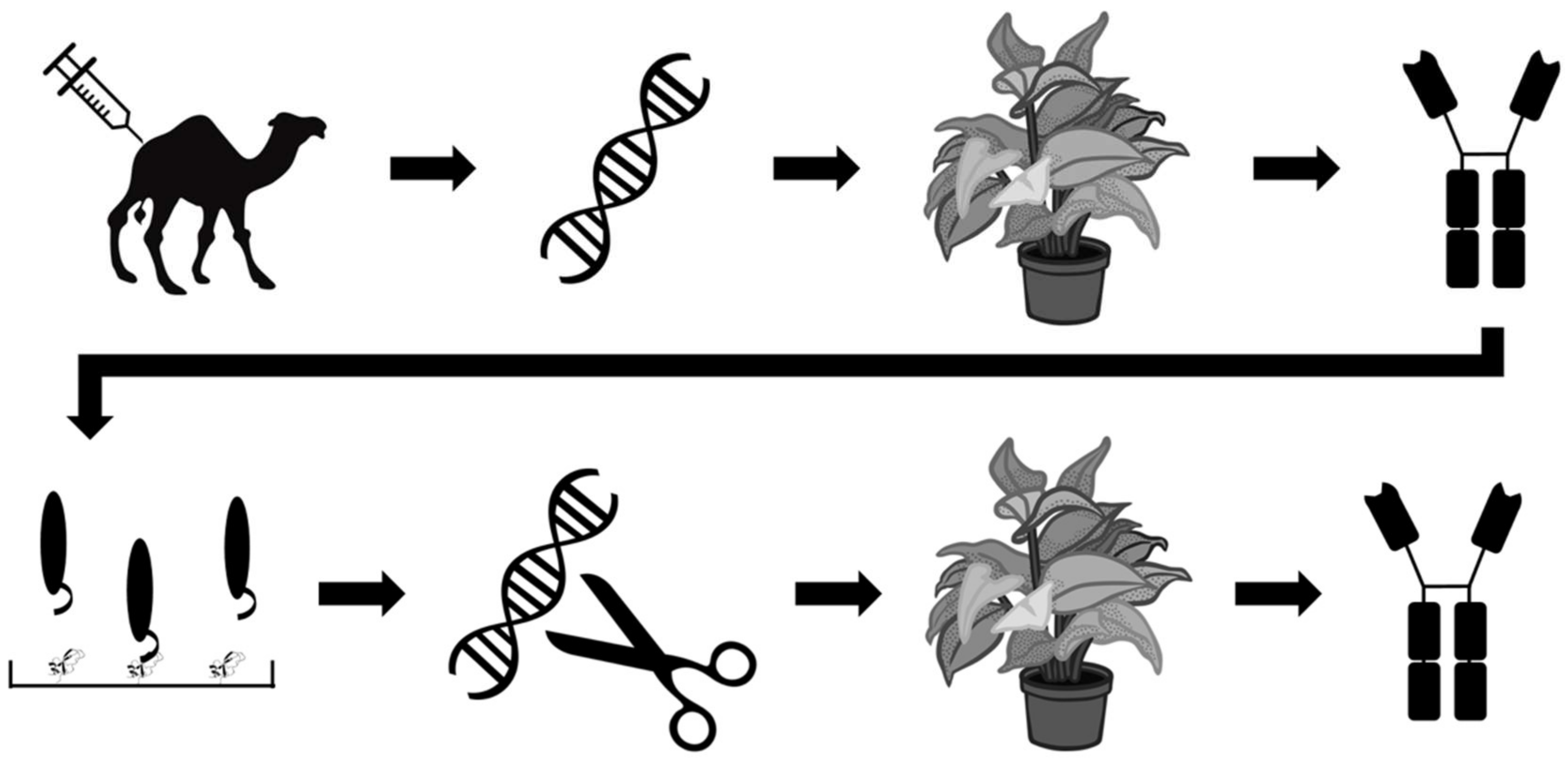
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.A.N.; Samarasekara, C.L.; Wagstaff, S.C.; Kinne, J.; Wernery, U.; Harrison, R.A. Analysis of camelid IgG for antivenom development: Immunoreactivity and preclinical neutralisation of venom-induced pathology by IgG subclasses, and the effect of heat treatment. Toxicon 2010, 56, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P.; Liu, J.H.; Zabetakis, D.; Liu, J.L.; Goldman, E.R. Thermal stabilization of anti-α-cobratoxin single domain antibodies. Toxicon 2017, 129, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Meyers, A.J.; McLean, M.D.; Arbabi-Ghahroudi, M.; MacKenzie, R.; Hall, J.C. In vivo neutralization of alpha-cobratoxin with high-affinity llama single-domain antibodies (VHHs) and a VHH-Fc antibody. PLoS ONE 2013, 8, e69495. [Google Scholar] [CrossRef] [PubMed]
- Julve Parreño, J.M.; Huet, E.; Fernández-del-Carmen, A.; Segura, A.; Venturi, M.; Gandía, A.; Pan, W.S.; Albaladejo, I.; Forment, J.; Pla, D.; et al. A synthetic biology approach for consistent production of plant-made recombinant polyclonal antibodies against snake venom toxins. Plant Biotechnol. J. 2018, 16, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Kuczewski, M.; Schirmer, E.; Lain, B.; Zarbis-Papastoitsis, G. A single-use purification process for the production of a monoclonal antibody produced in a PER.C6 human cell line. Biotechnol. J. 2011, 6, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, F.C.; Rasmussen, S.K.; Frandsen, T.P.; Rasmussen, L.K.; Tengbjerg, K.; Coljee, V.W.; Sharon, J.; Yang, C.-Y.; Bregenholt, S.; Nielsen, L.S.; et al. Production of target-specific recombinant human polyclonal antibodies in mammalian cells. Biotechnol. Bioeng. 2006, 94, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.K.; Næsted, H.; Müller, C.; Tolstrup, A.B.; Frandsen, T.P. Recombinant antibody mixtures: Production strategies and cost considerations. Arch. Biochem. Biophys. 2012, 526, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.J.; Hendriks, L.J.A.; De Kruif, J.; Throsby, M.; Heck, A.J.R. Complex mixtures of antibodies generated from a single production qualitatively and quantitatively evaluated by native Orbitrap mass spectrometry. MAbs 2014, 6, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Costing recombinant antivenoms. Nat. Corresp. 2016, 538, 41. [Google Scholar]
- Laustsen, A.H.; Johansen, K.H.; Engmark, M.R. Andersen recombinant snakebite antivenoms: A cost-effective solution to a neglected tropical disease? PLoS Negl. Trop. Dis. 2017, 11, e0005361. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G. Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 2014, 32, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Olinger, G.G.; Pettitt, J.; Kim, D.; Working, C.; Bohorov, O.; Bratcher, B.; Hiatt, E.; Hume, S.D.; Johnson, A.K.; Morton, J.; et al. Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc. Natl. Acad. Sci. USA 2012, 109, 18030–18035. [Google Scholar] [CrossRef] [PubMed]
- Murin, C.D.; Fusco, M.L.; Bornholdt, Z.A.; Qiu, X.; Olinger, G.G.; Zeitlin, L.; Kobinger, G.P.; Ward, A.B.; Saphire, E.O. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 2014, 111, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Juárez-González, V.R.; Riaño-Umbarila, L.; Quintero-Hernández, V.; Olamendi-Portugal, T.; Ortiz-León, M.; Ortíz, E.; Possani, L.D.; Becerril, B. Directed evolution, phage display and combination of evolved mutants: A strategy to recover the neutralization properties of the scFv version of BCF2 a neutralizing monoclonal antibody specific to scorpion toxin Cn2. J. Mol. Biol. 2005, 346, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Riaño-Umbarila, L.; Olamendi-Portugal, T.; Morelos-Juárez, C.; Gurrola, G.B.; Possani, L.D.; Becerril, B. A novel human recombinant antibody fragment capable of neutralizing Mexican scorpion toxins. Toxicon 2013, 76, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, E.R.; Olamendi-Portugal, T.; Serrano-Posada, H.; Arredondo-López, J.N.; Gómez-Ramírez, I.; Fernández-Taboada, G.; Possani, L.D.; Anguiano-Vega, G.A.; Riaño-Umbarila, L.; Becerril, B. Broadening the neutralizing capacity of a family of antibody fragments against different toxins from Mexican scorpions. Toxicon 2016, 119, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Espino-Solis, G.P.; Riaño-Umbarila, L.; Becerril, B.; Possani, L.D. Antidotes against venomous animals: State of the art and prospectives. J. Proteom. 2009, 72, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Funayama, J.C.; Pucca, M.B.; Roncolato, E.C.; Bertolini, T.B.; Campos, L.B.; Barbosa, J.E. Production of human antibody fragments binding to melittin and phospholipase A2 in Africanised bee venom: Minimising venom toxicity. Basic Clin. Pharmacol. Toxicol. 2012, 110, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Zoccal, K.F.; Roncolato, E.C.; Bertolini, T.B.; Campos, L.B.; Cologna, C.T.; Faccioli, L.H.; Arantes, E.C.; Barbosa, J.E. Serrumab: A human monoclonal antibody that counters the biochemical and immunological effects of tityus serrulatus venom. J. Immunotoxicol. 2012, 9, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Cerni, F.A.; Peigneur, S.; Arantes, E.C.; Tytgat, J.; Barbosa, J.E. Serrumab: A novel human single chain-fragment antibody with multiple scorpion toxin-neutralizing capacities. J. Immunotoxicol. 2014, 11, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Tamarozzi, M.B.; Soares, S.G.; Marcussi, S.; Giglio, J.R.; Barbosa, J.E. Expression of recombinant human antibody fragments capable of inhibiting the phospholipase and myotoxic activities of Bothrops jararacussu venom. Biochim. Biophys. Acta Gen. Subj. 2006, 1760, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.; Pucca, M.B.; Pessenda, G.; Campos, L.B.; Martinez, E.Z.; Cerni, F.A.; Barbosa, J.E. Discovery of human scFvs that cross-neutralize the toxic effects of B. jararacussu and C. d. terrificus venoms. Acta Trop. 2018, 177, 66–73. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knudsen, C.; Laustsen, A.H. Recent Advances in Next Generation Snakebite Antivenoms. Trop. Med. Infect. Dis. 2018, 3, 42. https://doi.org/10.3390/tropicalmed3020042
Knudsen C, Laustsen AH. Recent Advances in Next Generation Snakebite Antivenoms. Tropical Medicine and Infectious Disease. 2018; 3(2):42. https://doi.org/10.3390/tropicalmed3020042
Chicago/Turabian StyleKnudsen, Cecilie, and Andreas H. Laustsen. 2018. "Recent Advances in Next Generation Snakebite Antivenoms" Tropical Medicine and Infectious Disease 3, no. 2: 42. https://doi.org/10.3390/tropicalmed3020042
APA StyleKnudsen, C., & Laustsen, A. H. (2018). Recent Advances in Next Generation Snakebite Antivenoms. Tropical Medicine and Infectious Disease, 3(2), 42. https://doi.org/10.3390/tropicalmed3020042