
Development of a SYBR Green-Based Real-Time PCR Assay to Detect Oncomelania hupensis quadrasi DNA in Environmental Water Samples

 , ,
, ,  , , ,
, , , 

Abstract
1. Introduction
2. Methods
2.1. Ethical Considerations
2.2. Primer Design
2.3. PCR Controls
2.4. Water Samples from Snail Aquaria
2.5. Water Filtration and Sample Fixation
2.6. DNA Extraction and Quantification
2.7. Master Mix Preparation
2.8. Initial qPCR Conditions
2.9. Optimization of qPCR Reaction Conditions
2.9.1. Annealing Temperature
2.9.2. Optimal Primer Concentrations
2.9.3. Primer Stability
2.10. Assay Specificity, Sensitivity, and Efficiency
2.10.1. Assay Specificity
2.10.2. Assay Sensitivity
2.10.3. Assay Efficiency
2.11. Assay Validation Using Environmental Samples
2.12. Assay Diagnostic Performance
2.13. Study Design Limitation
3. Results
3.1. Primers
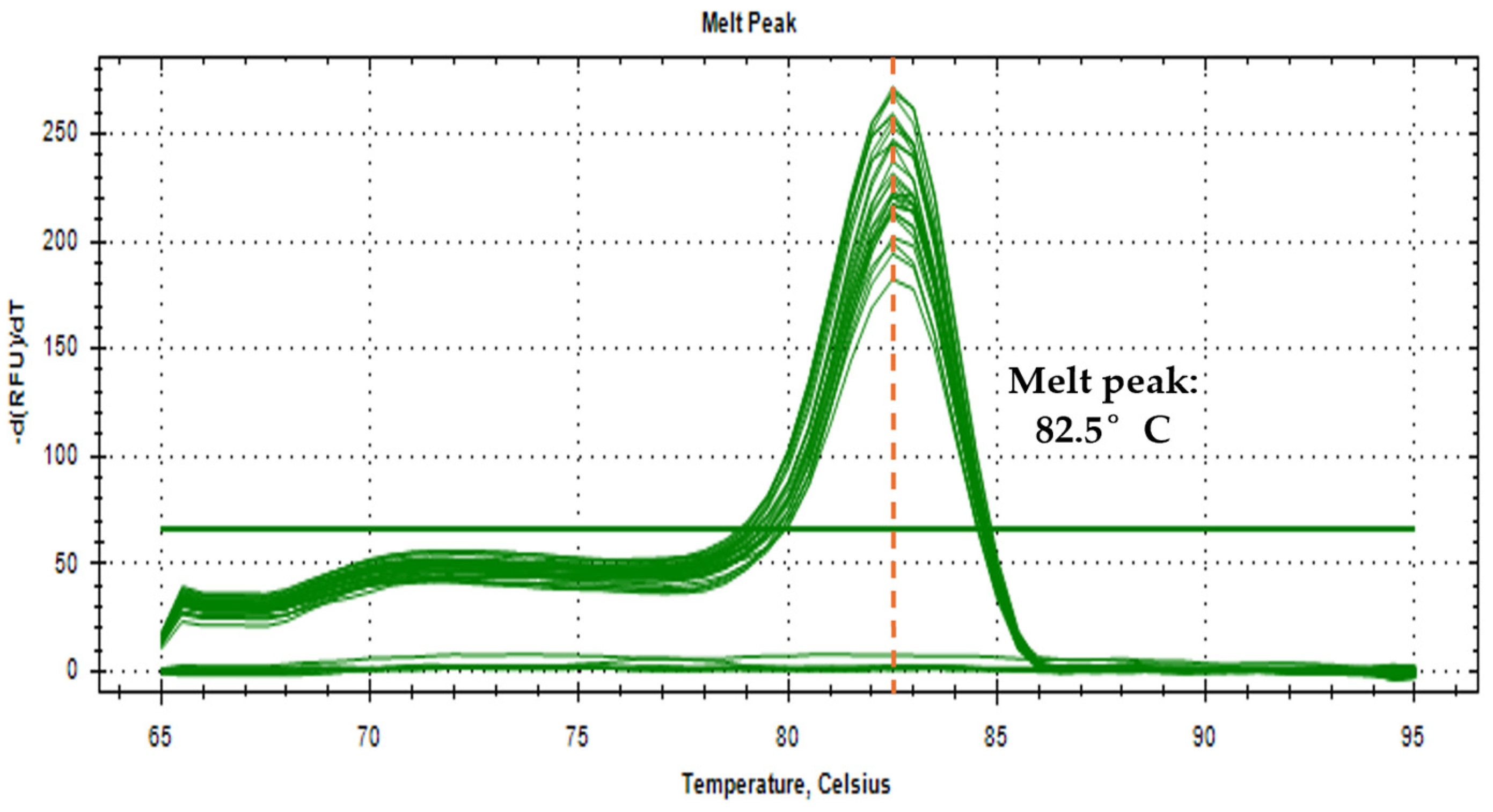
3.2. Optimization of qPCR Reaction Conditions
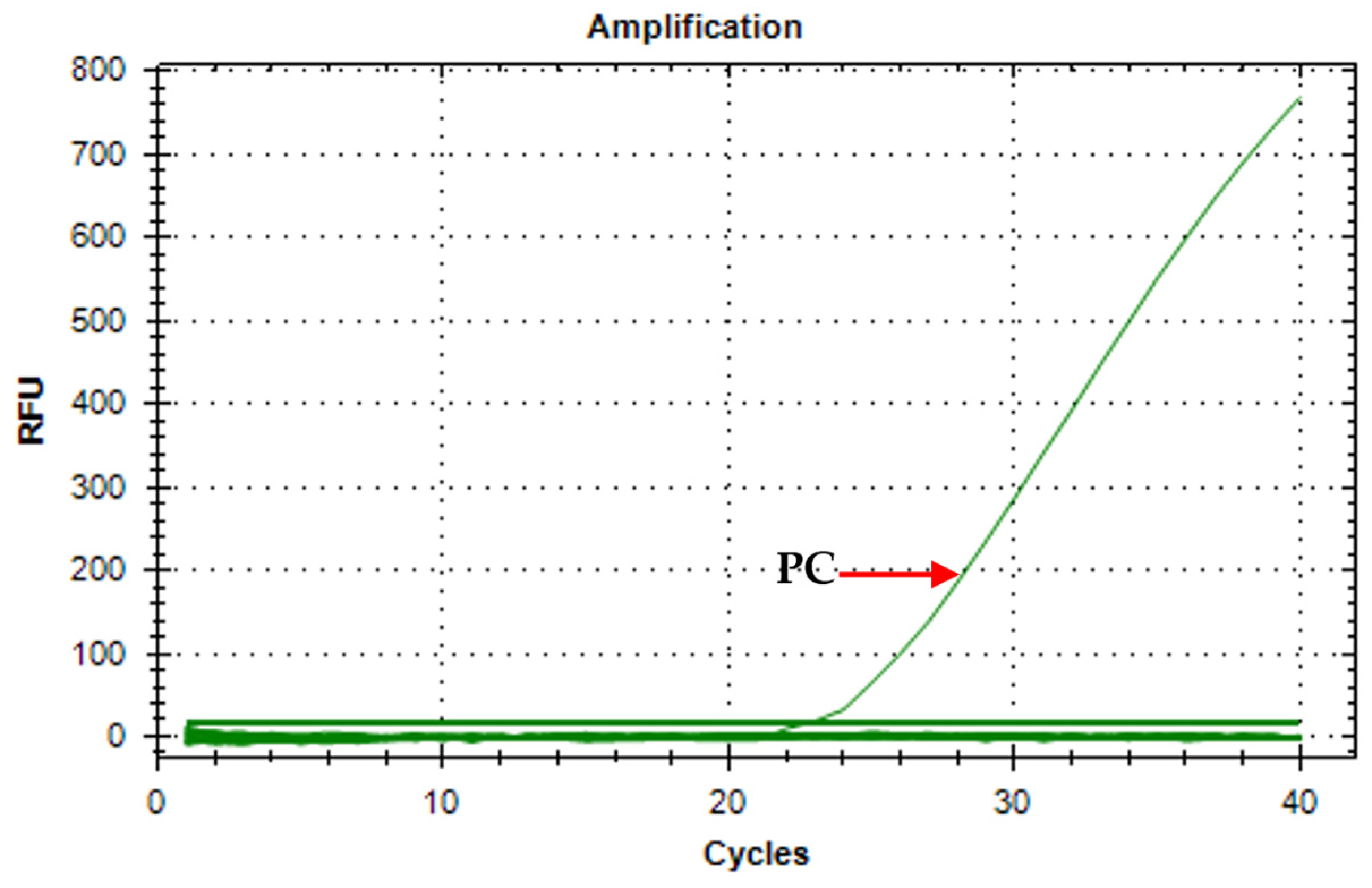
3.3. Assay Specificity
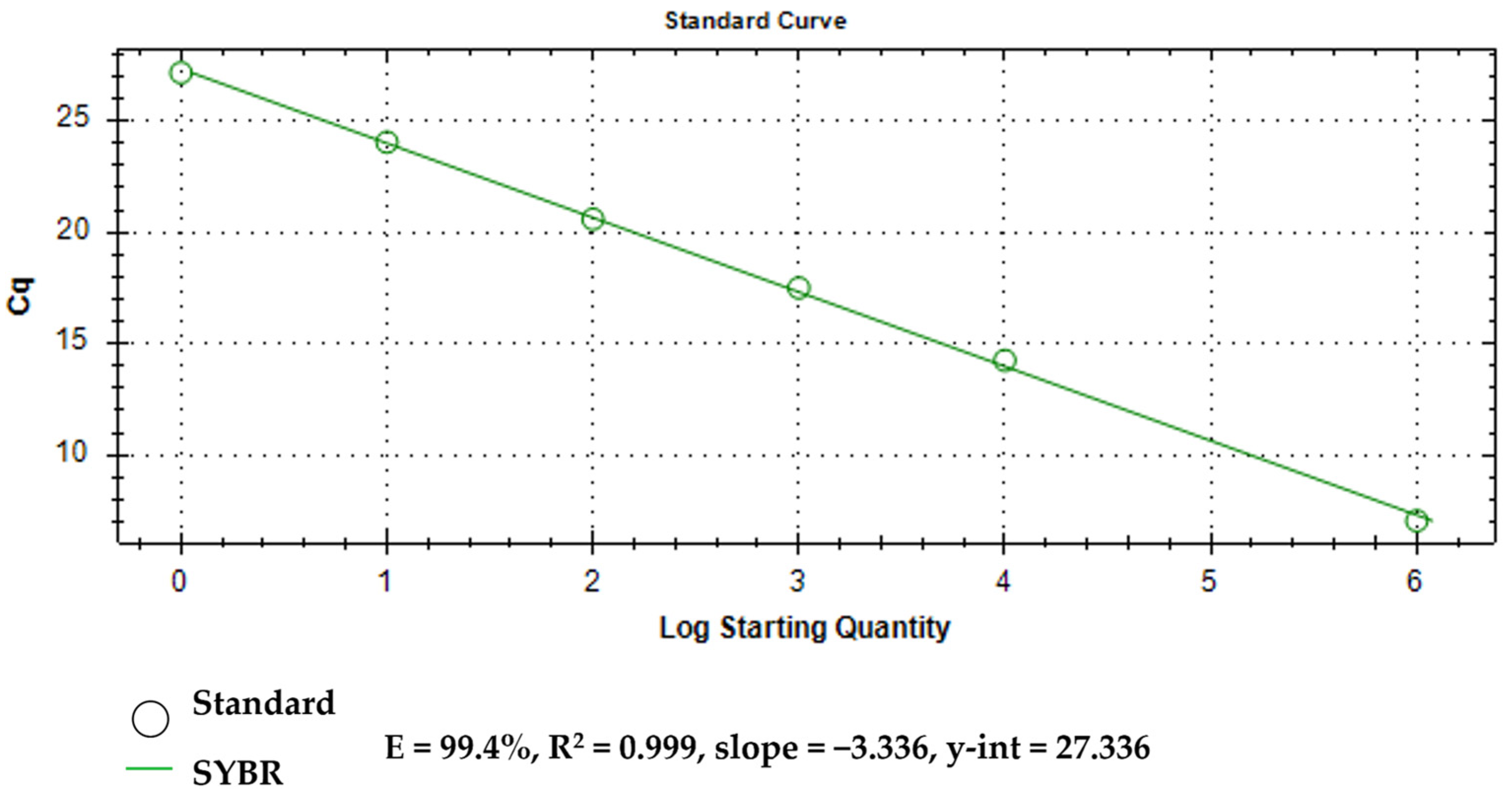
3.4. Assay Limit of Detection (LOD)
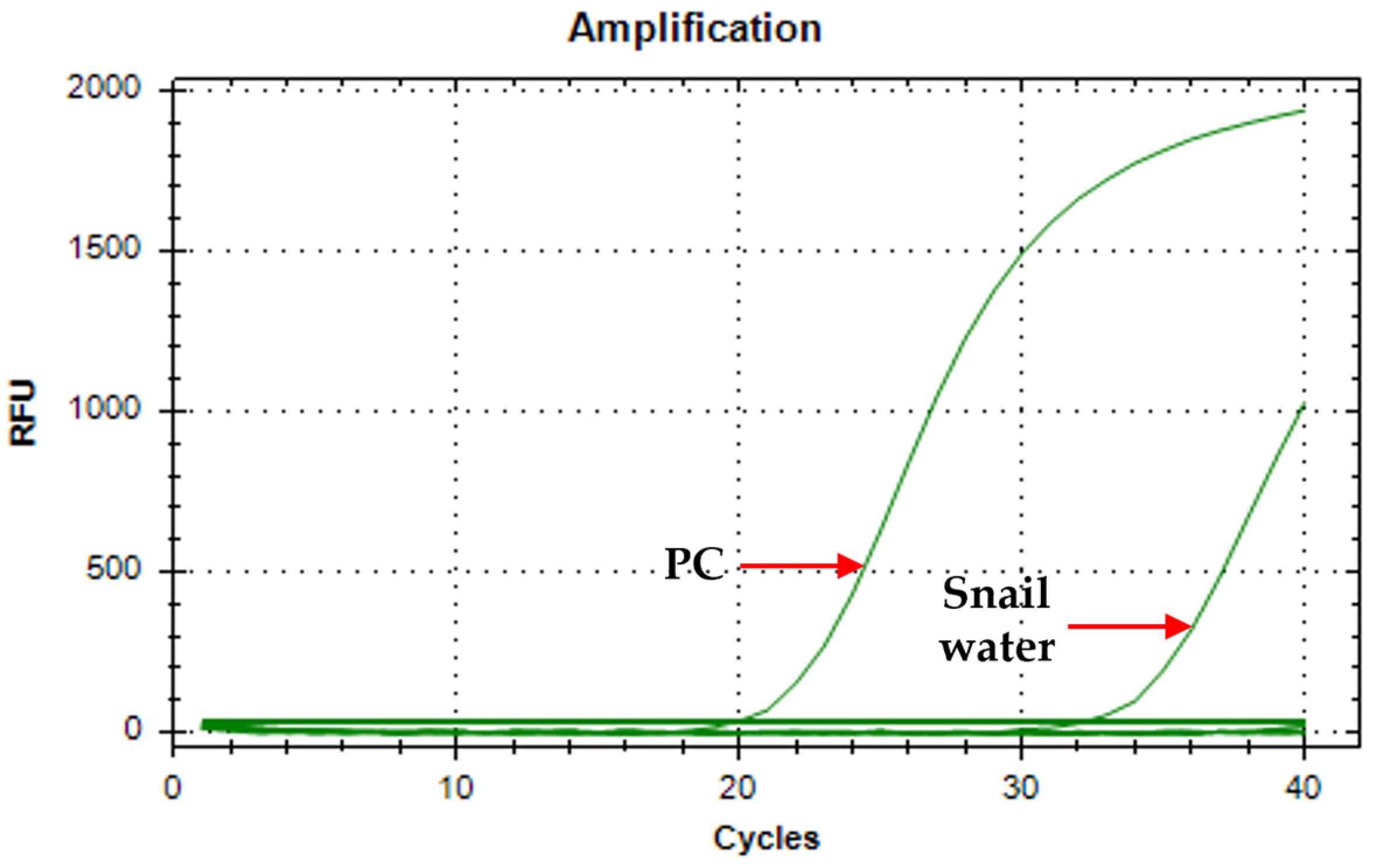
3.5. Assay Verification Using Environmental Waters
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olveda, R.M.; Tallo, V.; Olveda, D.U.; Inobaya, M.T.; Chau, T.N.; Ross, A.G. National survey data for zoonotic schistosomiasis in the Philippines grossly underestimates the true burden of disease within endemic zones: Implications for future control. Int. J. Infect. Dis. 2016, 45, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Fornillos, R.J.C.; Sato, M.O.; Tabios, I.K.B.; Sato, M.; Leonardo, L.R.; Chigusa, Y.; Minamoto, T.; Kikuchi, M.; Legaspi, E.R.; Fontanilla, I.K.C. Detection of Schistosoma japonicum and Oncomelania hupensis quadrasi environmental DNA and its potential utility to schistosomiasis japonica surveillance in the Philippines. PLoS ONE 2019, 14, e0224617. [Google Scholar] [CrossRef]
- Nelwan, M.L. Schistosomiasis: Life cycle, diagnosis, and control. Curr. Ther. Res. 2019, 91, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.C.; Molina, E. Schistosoma japonicum in the Philippines: Its epidemiology, diagnostics, control, and elimination. J. Agric. Res. Dev. Ext. Technology. 2021, 3, 71–87. [Google Scholar] [CrossRef]
- Leonardo, L.; Varona, G.; Fornillos, R.J.; Manalo, D.; Tabios, I.K.; Moendeg, K.; De Cadiz, A.; Kikuchi, M.; Chigusa, Y.; Mistica, M.; et al. Oncomelania hupensis quadrasi: Snail intermediate host of Schistosoma japonicum in the Philippines. Acta Trop. 2020, 210, 105547. [Google Scholar] [CrossRef]
- Leonardo, L.; Chigusa, Y.; Kikuchi, M.; Kato-Hayashi, N.; Kawazu, S.-I.; Angeles, J.; Fontanilla, I.; Tabios, I.; Moendeg, K.; Goto, Y.; et al. Review schistosomiasis in the Philippines: Challenges and some successes in control. Schistosomiasis Philipp. 2016, 47, 651–666. [Google Scholar]
- Wu, Y.; Liu, J.; Lin, Y.; Weng, R.; Chen, R.; Li, J.; Lv, Z. Diagnosis, Monitoring, and Control of Schistosomiasis—An update. J. Biomed. Nanotechnol. 2018, 14, 430–455. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, Z.; Dai, J.; Ke, Y.; Chen, X.; Hu, Y. Precision mapping of snail habitat in lake and marshland areas: Integrating environmental and textural indicators using Random Forest modeling. Heliyon 2024, 10, e36300. [Google Scholar] [CrossRef]
- Guan, W.; Li, S.; Abe, E.M.; Webster, B.L.; Rollinson, D.; Zhou, X. The genetic diversity and geographical separation study of Oncomelania hupensis populations in mainland China using microsatellite loci. Parasites Vectors 2016, 9, 28. [Google Scholar] [CrossRef]
- Hung, Y.W.; Remais, J. Quantitative Detection of Schistosoma japonicum Cercariae in Water by Real-Time PCR. PLoS Neglected Trop. Dis. 2008, 2, e337. [Google Scholar] [CrossRef]
- Tajadini, M.; Panjehpour, M.; Javanmard, S. Comparison of SYBR Green and TaqMan methods in quantitative real-time polymerase chain reaction analysis of four adenosine receptor subtypes. Adv. Biomed. Res. 2014, 3, 85. [Google Scholar] [CrossRef]
- Ficetola, G.F.; Poulenard, J.; Sabatier, P.; Messager, E.; Gielly, L.; Leloup, A.; Etienne, D.; Bakke, J.; Malet, E.; Fanget, B.; et al. DNA from lake sediments reveals long-term ecosystem changes after a biological invasion. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, T.; Yamanaka, H.; Takahara, T.; Honjo, M.N.; Kawabata, Z. Surveillance of fish species composition using environmental DNA. Limnology 2011, 13, 193–197. [Google Scholar] [CrossRef]
- Hashizume, H.; Sato, M.; Sato, M.O.; Ikeda, S.; Yoonuan, T.; Sanguankiat, S.; Pongvongsa, T.; Moji, K.; Minamoto, T. Application of environmental DNA analysis for the detection of Opisthorchis viverrini DNA in water samples. Acta Trop. 2017, 169, 1–7. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Willerslev, E. Environmental DNA—An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Worrell, C.; Xiao, N.; Vidal, J.E.; Chen, L.; Zhong, B.; Remais, J. Field Detection of Schistosoma japonicum Cercariae in Environmental Water Samples by Quantitative PCR. Appl. Environ. Microbiol. 2011, 77, 2192–2195. [Google Scholar] [CrossRef]
- Calata, F.I.C.; Caranguian, C.Z.; Mendoza, J.E.M.; Fornillos, R.J.C.; Tabios, I.K.B.; Fontanilla, I.K.C.; Leonardo, L.R.; Sunico, L.S.; Kawai, S.; Chigusa, Y.; et al. Analysis of Environmental DNA and Edaphic Factors for the Detection of the Snail Intermediate Host Oncomelania hupensis quadrasi. Pathogens 2019, 8, 160. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Zhao, F.; Maren, N.A.; Kosentka, P.Z.; Liao, Y.-Y.; Lu, H.; Duduit, J.R.; Huang, D.; Ashrafi, H.; Zhao, T.; Huerta, A.I.; et al. An optimized protocol for stepwise optimization of real-time RT-PCR analysis. Hortic. Res. 2021, 8, 179. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Schoonjans, F. MedCalc’s Diagnostic Test Evaluation Calculator. MedCalc. Available online: https://www.medcalc.org/calc/diagnostic_test.php (accessed on 9 October 2023).
- Why do I Have Multiple Peaks in My Melt Curve? (n.d.). New England Biolabs. Available online: https://www.neb.com/en/faqs/2016/11/14/why-do-i-have-multiple-peaks-in-my-melt-curve#:~:text=The%20presence%20of%20multiple%20DNA,or%20off%2Dtarget%20amplification%20products (accessed on 7 April 2025).
- Madsen, H.; Carabin, H.; Balolong, D.; Tallo, V.L.; Olveda, R.; Yuan, M.; McGarvey, S.T. Prevalence of Schistosoma japonicum infection of Oncomelania quadrasi snail colonies in 50 irrigated and rain-fed villages of Samar Province, the Philippines. Acta Trop. 2007, 105, 235–241. [Google Scholar] [CrossRef]
- Lv, C.; Li, Y.; Deng, W.; Bao, Z.; Xu, J.; Lv, S.; Li, S.; Zhou, X. The Current Distribution of Oncomelania hupensis Snails in the People’s Republic of China Based on a Nationwide Survey. Trop. Med. Infect. Dis. 2023, 8, 120. [Google Scholar] [CrossRef]
- Hyndman, D.L.; Mitsuhashi, M. PCR primer design. Methods Mol. Biol. 2003, 226, 81–88. [Google Scholar]
- Rodríguez, A.; Rodríguez, M.; Córdoba, J.J.; Andrade, M.J. Design of primers and probes for quantitative real-time PCR methods. In PCR Primer Design; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1275, pp. 31–56. [Google Scholar]
- Raymaekers, M.; Smets, R.; Maes, B.; Cartuyvels, R. Checklist for optimization and validation of real-time PCR assays. J. Clin. Lab. Anal. 2009, 23, 145–151. [Google Scholar] [CrossRef]
- Wu, J.Y.; Jiang, X.T.; Jiang, Y.X.; Lu, S.Y.; Zou, F.; Zhou, H.W. Effects of polymerase, template dilution and cycle number on PCR based 16 S rRNA diversity analysis using the deep sequencing method. BMC Microbiol. 2010, 10, 255. [Google Scholar] [CrossRef]
- Kumar, A.; Kaur, J. Primer Based Approach for PCR Amplification of High GC Content Gene: Mycobacterium Gene as a Model. Mol. Biol. Int. 2014, 937308. [Google Scholar]
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019, 12, 106. [Google Scholar]
- DNA Amplification|Thermal Cycling Parameters & Optimization|National Institute of Justice. (n.d.). National Institute of Justice. Available online: http://nij.ojp.gov/nij-hosted-online-training-courses/dna-amplification/overview/thermal-cycling-parameters-optimization (accessed on 22 July 2024).
- Bolin, E.; Lam, W. A review of sensitivity, specificity, and likelihood ratios: Evaluating the utility of the electrocardiogram as a screening tool in hypertrophic cardiomyopathy. Congenit. Heart Dis. 2013, 8, 406–410. [Google Scholar] [CrossRef]
- Xia, Z.; Johansson, M.L.; Gao, Y.; Zhang, L.; Haffner, G.D.; MacIsaac, H.J.; Zhan, A. Conventional versus real-time quantitative PCR for rare species detection. Ecol. Evol. 2018, 8, 11799–11807. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Bustin, S.A. (Ed.) A-Z of Quantitative PCR; International University Line: La Jolla, CA, USA, 2004. [Google Scholar]
- Navidshad, B.; Liang, J.B.; Jahromi, M.F. Correlation coefficients between different methods of expressing bacterial quantification using real time PCR. Int. J. Mol. Sci. 2012, 13, 2119–2132. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Length (bp) | Tm (°C) | %GC |
|---|---|---|---|---|
| Forward | 5′-CCAGCGCAAAGCTCGTTTAG-3′ | 20 | 60.18 | 55 |
| Reverse | 5′-TCAAGAGAATCTCCAGGCACG-3′ | 21 | 59.79 | 52 |
| Annealing Temperature (°C) | No-Template Control | Ohq Positive Control (Ct Values) | ||||
|---|---|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | Mean | Standard Deviation | ||
| 56.0 | Invalid | 20.59 | 21.11 | 21.07 | 20.92 | 0.289 |
| 56.6 | Invalid | 20.53 | 20.96 | 21.22 | 20.90 | 0.348 |
| 57.8 | Valid | 20.61 | 20.74 | 21.6 | 20.98 | 0.538 |
| 59.6 | Valid | 19.51 | 19.06 | 20.11 | 19.56 | 0.527 |
| 61.7 | Valid | 19.63 | 19.83 | 19.65 | 19.70 | 0.110 |
| 63.0 | Valid | 19.51 | 19.53 | 19.47 | 19.50 | 0.031 |
| 64.5 | Valid | 19.67 | 19.65 | 19.83 | 19.72 | 0.099 |
| 65.0 | Valid | 19.51 | 19.62 | 19.61 | 19.58 | 0.061 |
| Primer Concentration (µM) | No-Template Control | Ohq Positive Control (Ct Values) | ||||
|---|---|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | Mean | Standard Deviation | ||
| 1.0 | Valid | 31.11 | 30.09 | 30.26 | 30.49 | 0.546 |
| 2.0 | Valid | 21.49 | 21.78 | 21.39 | 21.55 | 0.202 |
| 5.0 | Valid | 19.81 | 19.67 | 19.55 | 19.68 | 0.130 |
| 10.0 | Valid | 19.52 | 19.27 | 19.02 | 19.27 | 0.250 |
| Snail Species | Ct Value | ||
|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | |
| Oncomelania hupensis quadrasi | 19.37 | 19.44 | 19.41 |
| Jagora asperata | NA | NA | NA |
| Vivipara zamboagensis surigensis | NA | NA | NA |
| Radix quadrasi | NA | NA | NA |
| Pomacea canaliculata | NA | NA | NA |
| No-template control | Valid | Valid | Valid |
| Mean Concentration (Copies/µL) | % Replicate Detection N = 8 | Mean Ct | Ct SD (≥95% CI) |
|---|---|---|---|
| 106 | 100 (8/8) | 7.14 | 0.18 |
| 105 | 100 (8/8) | 14.57 | 0.50 |
| 104 | 100 (8/8) | 17.63 | 0.15 |
| 103 | 100 (8/8) | 21.03 | 0.18 |
| 102 | 100 (8/8) | 24.21 | 0.11 |
| 101 | 100 (8/8) | 26.84 | 0.30 |
| 100 | 100 (8/8) | 28.78 | 1.01 |
| 10−1 | 100 (7/8) | 30.79 | 1.34 |
| 10−2 | 100 (7/8) | 31.47 | 1.39 |
| No-template control | 0 (0/8) | NA | NA |
| Ohq-Infected Environmental Water Sample | |||
|---|---|---|---|
| Positive | Negative | ||
| Ohq qPCR assay detection | Positive | 30 | 0 |
| Negative | 0 | 30 | |
| Performance Criteria | Estimated Value (%) | 95% CI (%) |
|---|---|---|
| Positive Predictive Value (PPV) | 100 | 88.43 to 100.00 |
| Negative Predictive Value (NPV) | 100 | 88.43 to 100.00 |
| Computed using: https://www.medcalc.org/calc/diagnostic_test.php (accessed on 27 March 2025) [21] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manalo, D.L.; Bolivar, J.K.G.; Ermino, K.I.T.; Bondoc, J.G.; Espino, M.J.M.; Panganiban, E.P.; Nogrado, K.S.; Fornillos, R.J.C.; Jiz, M.A.; Leonardo, L.R.; et al. Development of a SYBR Green-Based Real-Time PCR Assay to Detect Oncomelania hupensis quadrasi DNA in Environmental Water Samples. Trop. Med. Infect. Dis. 2025, 10, 140. https://doi.org/10.3390/tropicalmed10050140
Manalo DL, Bolivar JKG, Ermino KIT, Bondoc JG, Espino MJM, Panganiban EP, Nogrado KS, Fornillos RJC, Jiz MA, Leonardo LR, et al. Development of a SYBR Green-Based Real-Time PCR Assay to Detect Oncomelania hupensis quadrasi DNA in Environmental Water Samples. Tropical Medicine and Infectious Disease. 2025; 10(5):140. https://doi.org/10.3390/tropicalmed10050140
Chicago/Turabian StyleManalo, Daria L., Jude Karlo G. Bolivar, Karl Ian T. Ermino, Jeromir G. Bondoc, Mark Joseph M. Espino, Efraim P. Panganiban, Kathyleen S. Nogrado, Raffy Jay C. Fornillos, Mario A. Jiz, Lydia R. Leonardo, and et al. 2025. "Development of a SYBR Green-Based Real-Time PCR Assay to Detect Oncomelania hupensis quadrasi DNA in Environmental Water Samples" Tropical Medicine and Infectious Disease 10, no. 5: 140. https://doi.org/10.3390/tropicalmed10050140
APA StyleManalo, D. L., Bolivar, J. K. G., Ermino, K. I. T., Bondoc, J. G., Espino, M. J. M., Panganiban, E. P., Nogrado, K. S., Fornillos, R. J. C., Jiz, M. A., Leonardo, L. R., & Fontanilla, I. K. C. (2025). Development of a SYBR Green-Based Real-Time PCR Assay to Detect Oncomelania hupensis quadrasi DNA in Environmental Water Samples. Tropical Medicine and Infectious Disease, 10(5), 140. https://doi.org/10.3390/tropicalmed10050140