
Discovery of Targetable Epitopes in Tomato Chlorosis Virus Through Comparative Genomics and Structural Modeling


Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Whole Genomes
2.2. Comparative Genomic Analysis and Phylogenic Analysis
2.3. Cellular Localization Prediction
2.4. Structure Assessment and Epitope Prediction
3. Results
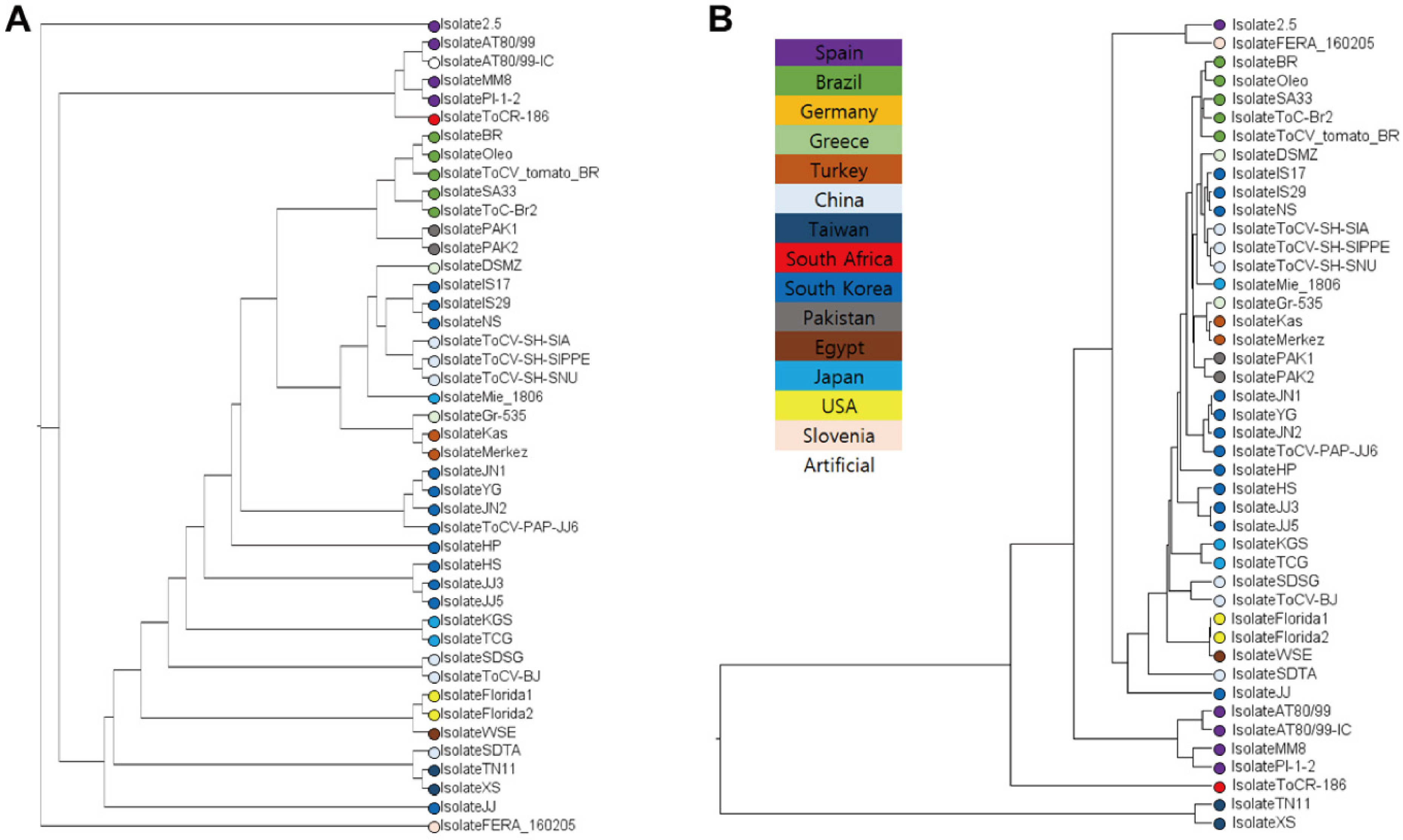
3.1. Taiwanese Isolates Form the Most Distinct Clade Among ToCV Populations
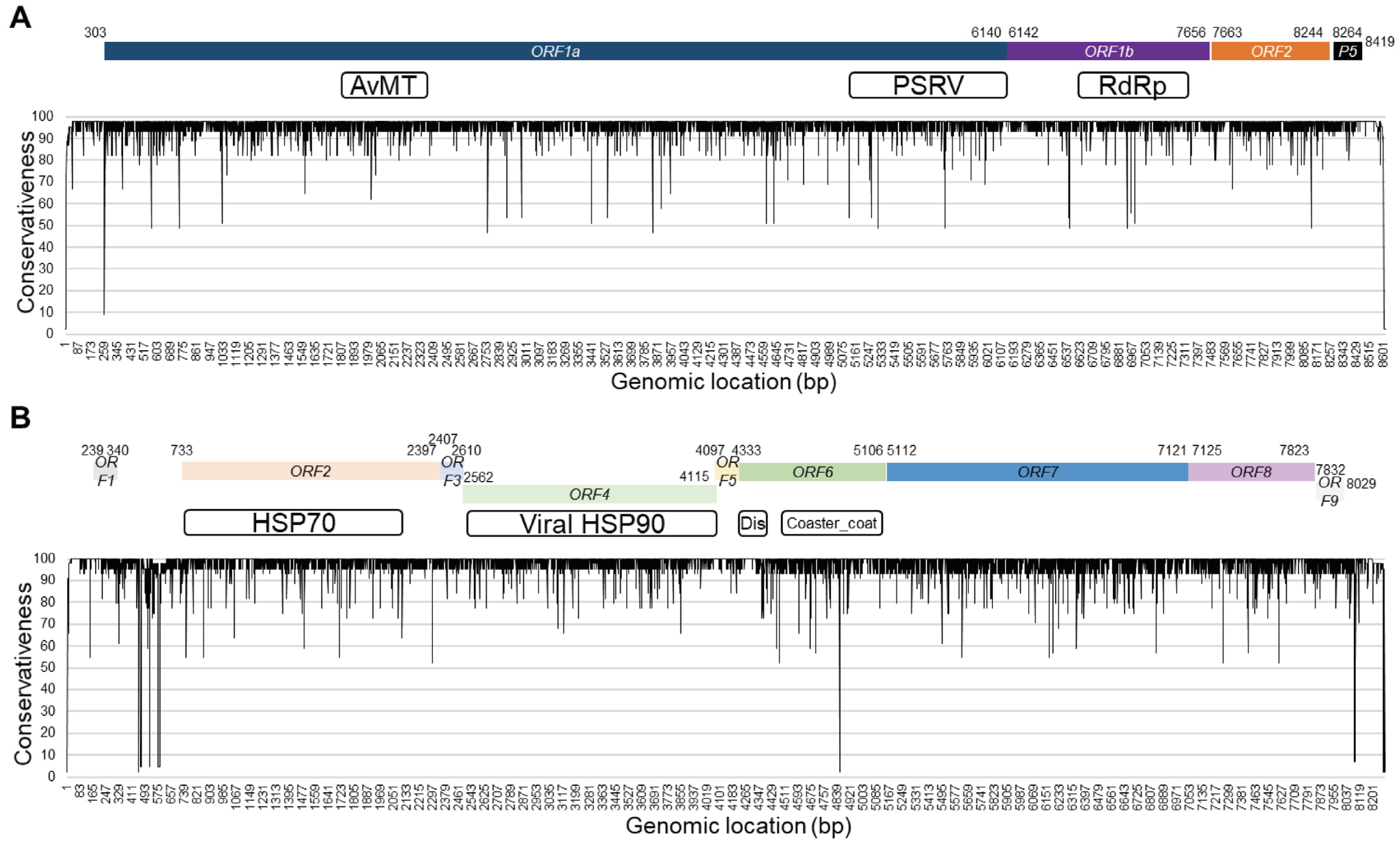
3.2. Coding Regions of the ToCV Genome Exhibit High Sequence Conservation
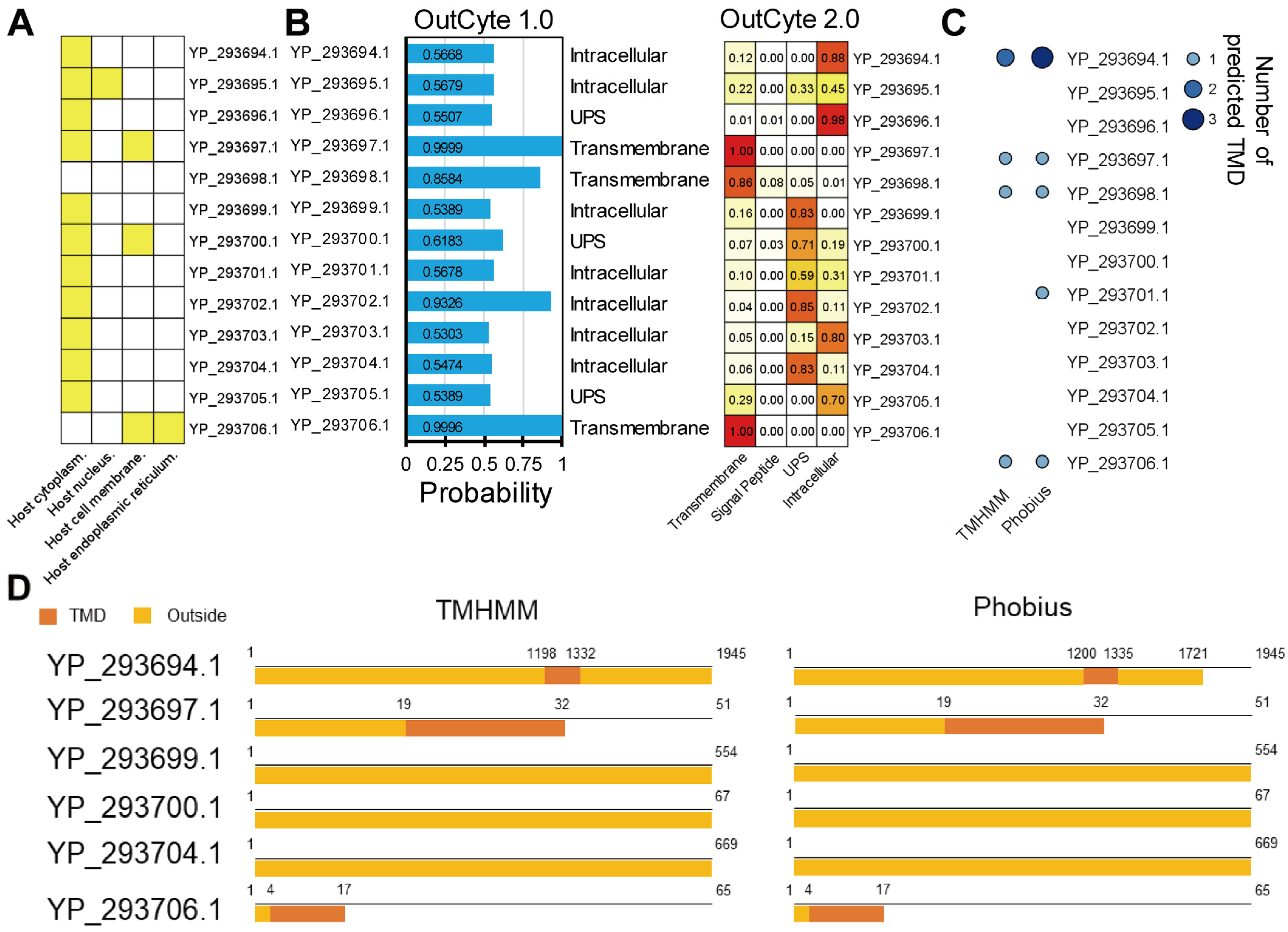
3.3. Six ToCV Proteins Are Predicted to Contain Peptide Regions Exposed on the Outer Side of Plant Cells
3.4. Four Highly Probable Epitopes Are Identified
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, an emergent plant virus still expanding its geographical and host ranges. Mol. Plant Pathol. 2019, 20, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Fortes, I.M.; Moriones, E.; Navas-Castillo, J. Tomato chlorosis virus in pepper: Prevalence in commercial crops in southeastern Spain and symptomatology under experimental conditions. Plant Pathol. 2012, 61, 994–1001. [Google Scholar] [CrossRef]
- Lozano, G.; Moriones, E.; Navas-Castillo, J. Complete sequence of the RNA1 of a European isolate of tomato chlorosis virus. Arch. Virol. 2007, 152, 839–841. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Moriones, E.; Navas-Castillo, J. Complete nucleotide sequence of the RNA2 of the crinivirus tomato chlorosis virus. Arch. Virol. 2006, 151, 581–587. [Google Scholar] [CrossRef]
- Wintermantel, W.M.; Wisler, G.C.; Anchieta, A.G.; Liu, H.-Y.; Karasev, A.V.; Tzanetakis, I.E. The complete nucleotide sequence and genome organization of Tomato chlorosis virus. Arch. Virol. 2005, 150, 2287–2298. [Google Scholar] [CrossRef]
- Chen, A.Y.S.; Walker, G.P.; Carter, D.; Ng, J.C.K. A virus capsid component mediates virion retention and transmission by its insect vector. Proc. Natl. Acad. Sci. USA 2011, 108, 16777–16782. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kil, E.J.; Kwak, H.R.; Kim, M.; Seo, J.K.; Lee, S.; Choi, H.S. Phylogenetic Characterization of Tomato chlorosis virus Population in Korea: Evidence of Reassortment between Isolates from Different Origins. Plant Pathol. J. 2018, 34, 199–207. [Google Scholar] [CrossRef]
- Orfanidou, C.G.; Pappi, P.G.; Efthimiou, K.E.; Katis, N.I.; Maliogka, V.I. Transmission of Tomato chlorosis virus (ToCV) by Bemisia tabaci Biotype Q and Evaluation of Four Weed Species as Viral Sources. Plant Dis. 2016, 100, 2043–2049. [Google Scholar] [CrossRef]
- Snopkowska, L.; Sara, W.; Maschio, D.; Henriquez-Camacho, C.; Moreno, C.V. Biomarkers for SARS-CoV-2 infection. A narrative review. Front. Med. 2025, 12, 1563998. [Google Scholar]
- Joshi, K.M.; Salve, S.; Dhanwade, D.; Chavhan, M.; Jagtap, S.; Shinde, M.; Holkar, R.; Patil, R.; Chabukswar, V. Advancing protein biosensors: Redefining detection through innovations in materials, mechanisms, and applications for precision medicine and global diagnostics. RSC Adv. 2025, 15, 11523–11536. [Google Scholar] [CrossRef]
- Tu, L.; Wu, S.; Gan, S.; Zhao, W.; Li, S.; Cheng, Z.; Zhou, Y.; Zhu, Y.; Ji, Y. A simplified RT-PCR assay for the simultaneous detection of tomato chlorosis virus and tomato yellow leaf curl virus in tomato. J. Virol. Methods 2022, 299, 114282. [Google Scholar] [CrossRef] [PubMed]
- Morellos, A.; Tziotzios, G.; Orfanidou, C.; Pantazi, X.E.; Sarantaris, C.; Maliogka, V.; Alexandridis, T.K.; Moshou, D. Non-Destructive Early Detection and Quantitative Severity Stage Classification of Tomato Chlorosis Virus (ToCV) Infection in Young Tomato Plants Using Vis–NIR Spectroscopy. Remote Sens. 2020, 12, 1920. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Drummond, A.J.; Nicholls, G.K.; Rodrigo, A.G.; Solomon, W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 2002, 161, 1307–1320. [Google Scholar] [CrossRef]
- Barba-Montoya, J.; Tao, Q.; Kumar, S. Using a GTR+Γ substitution model for dating sequence divergence when stationarity and time-reversibility assumptions are violated. Bioinformatics 2020, 36 (Suppl. S2), i884–i894. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. Virus-mPLoc: A fusion classifier for viral protein subcellular location prediction by incorporating multiple sites. J. Biomol. Struct. Dyn. 2010, 28, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Poschmann, G.; Waldera-Lupa, D.; Rafiee, N.; Kollmann, M.; Stühler, K. OutCyte: A novel tool for predicting unconventional protein secretion. Sci. Rep. 2019, 9, 19448. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Song, W.; Luo, G.; Jiang, T. EpiScan: Accurate high-throughput mapping of antibody-specific epitopes using sequence information. Npj Syst. Biol. Appl. 2024, 10, 101. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Yang, F.; Leng, C.; Shen, X.; Bagas, L.; Zhang, L.; Jepson, G. Editorial: Evolution of tectonic structures and mineralisation in orogens and their margins. Front. Earth Sci. 2024, 12, 1371835. [Google Scholar] [CrossRef]
- Kanakala, S.; Ghanim, M. Global genetic diversity and geographical distribution of Bemisia tabaci and its bacterial endosymbionts. PLoS ONE 2019, 14, e0213946. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.S.; Snyder, W.E. What Is the Spatial Extent of a Bemisia tabaci Population? Insects 2020, 11, 813. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.G.; Newton, A.S.; Estrada, D. Intraplant and Interplant Movement of Bemisia argentifolii (Homoptera: Aleyrodidae) Crawlers. Environ. Entomol. 1996, 25, 1360–1364. [Google Scholar] [CrossRef]
- Byrne, D.N. Migration and dispersal by the sweet potato whitefly, Bemisia tabaci. Agric. For. Meteorol. 1999, 97, 309–316. [Google Scholar] [CrossRef]
- Shang, K.; Xiao, L.; Zhang, X.; Zang, L.; Zhao, D.; Wang, C.; Wang, X.; Zhou, T.; Zhu, C.; Zhu, X. Tomato chlorosis virus p22 interacts with NbBAG5 to inhibit autophagy and regulate virus infection. Mol. Plant Pathol. 2023, 24, 425–435. [Google Scholar] [CrossRef]
- Feng, L.; Luo, X.; Huang, L.; Zhang, Y.; Li, F.; Li, S.; Zhang, Z.; Yang, X.; Wang, X.; OuYang, X.; et al. A viral protein activates the MAPK pathway to promote viral infection by downregulating callose deposition in plants. Nat. Commun. 2024, 15, 10548. [Google Scholar] [CrossRef]
- Shi, X.; Yue, H.; Wei, Y.; Preisser, E.L.; Wang, P.; Du, J.; Xia, J.; Li, K.; Yang, X.; Chen, J.; et al. Neophytadiene, a Plant Specialized Metabolite, Mediates the Virus-Vector-Plant Tripartite Interactions. Adv. Sci. 2025, 12, 2416891. [Google Scholar] [CrossRef]
- Sun, X.; Zang, L.; Liu, X.; Jiang, S.; Zhang, X.; Zhao, D.; Shang, K.; Zhou, T.; Zhu, C.; Zhu, X. Interactions of Tomato Chlorosis Virus p27 Protein with Tomato Catalase Are Involved in Viral Infection. Viruses 2023, 15, 990. [Google Scholar] [CrossRef]
- Navas-Hermosilla, E.; Fiallo-Olivé, E.; Navas-Castillo, J. Infectious Clones of Tomato Chlorosis Virus: Toward Increasing Efficiency by Introducing the Hepatitis Delta Virus Ribozyme. Front. Microbiol. 2021, 12, 693457. [Google Scholar] [CrossRef]
- Kiss, Z.A.; Medina, V.; Falk, B. Crinivirus replication and host interactions. Front. Microbiol. 2013, 4, 99. [Google Scholar] [CrossRef]
- Kim, J.Y.; Symeonidi, E.; Pang, T.Y.; Denyer, T.; Weidauer, D.; Bezrutczyk, M.; Miras, M.; Zöllner, N.; Hartwig, T.; Wudick, M.M.; et al. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell 2021, 33, 511–530. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Geo_Location | Host | RNA1 | RNA2 | Collection_Date |
|---|---|---|---|---|---|
| 2.5 | Spain, Malaga | Solanum lycopersicum | KJ200304.1 | KJ200305.1 | 2010 |
| AT80/99 | Spain, Malaga | Solanum lycopersicum | DQ983480.1 | DQ136146.1 | 1999 |
| AT80/99-IC | Spain, Malaga | Solanum lycopersicum | KJ740256.1 | KJ740257.1 | 2014 |
| BR | Brazil, Sumaré, São Paulo | Capsicum annuum | MT279194.1 | MT279195.1 | 2019 |
| DSMZ PV-1242 | Greece | Solanum lycopersicum | ON398512.1 | ON398513.1 | 2018-10-02 |
| Gr-535 | Greece | not applicable | EU284745.1 | EU284744.1 | 2008 |
| HP | Republic of Korea | Solanum lycopersicum | KP114530.1 | KP114537.1 | 2013-01-01 |
| HS | Republic of Korea | Solanum lycopersicum | KP137098.1 | KP137099.1 | 2013-01-01 |
| IS17 | Republic of Korea | Solanum lycopersicum | KP114535.1 | KP114525.1 | 2013-01-01 |
| IS29 | Republic of Korea | Solanum lycopersicum | KP114538.1 | KP114529.1 | 2013-01-01 |
| JJ | Republic of Korea | Solanum lycopersicum | KP137100.1 | KP137101.1 | 2013-01-01 |
| JJ3 | Republic of Korea | not applicable | KP114532.1 | KP114533.1 | 2013-01-01 |
| JJ5 | Republic of Korea | Solanum lycopersicum | KP114527.1 | KP114534.1 | 2013-01-01 |
| JN1 | Republic of Korea | not applicable | KP114531.1 | KP114536.1 | 2013-01-01 |
| JN2 | Republic of Korea | Solanum lycopersicum | MG813909.1 | MG813910.1 | 2013 |
| Kas | Turkey | Solanum lycopersicum | KY419526.1 | KY419528.1 | 2015-02-03 |
| Merkez | Turkey | Solanum lycopersicum | KY419525.1 | KY419527.1 | 2015-02-03 |
| MM8 | Spain, Malaga | Capsicum annuum | KJ200306.1 | KJ200307.1 | 2005 |
| NS | Republic of Korea | Solanum lycopersicum | MG813908.1 | MG813911.1 | 2013 |
| Oleo | Brazil | Cucumis sativus | MN172419.1 | MN172420.1 | 2019-02-01 |
| PAK1 | Pakistan | Solanum lycopersicum | MN869004.1 | MN869006.1 | 2019-02-13 |
| PAK2 | Pakistan | Solanum lycopersicum | MN869005.1 | MN869007.1 | 2019-02-14 |
| Pl-1-2 | Spain, Malaga | Solanum lycopersicum | KJ200308.1 | KJ200309.1 | 1997 |
| SA33 | Brazil, Santo Amaro da Imperatriz, Santa Catarina | Solanum lycopersicum | OR283247.1 | OR283248.1 | 2020-08-01 |
| SDSG | China | Solanum lycopersicum | KC709509.1 | KC709510.1 | 2012-10-01 |
| SDTA | China | Solanum lycopersicum | OR246919.1 | OR246920.1 | 2019-01-09 |
| TN11 | Taiwan | Solanum lycopersicum | MF795556.1 | MF795557.1 | 1998 |
| ToC-Br2 | Brazil, Sao Paulo | Solanum lycopersicum | JQ952600.1 | JQ952601.1 | 2006 |
| ToCR-186 | South Africa | Solanum lycopersicum | KY471129.1 | KY471130.1 | 2015-02-15 |
| ToCV_tomato_BR | Brazil | Solanum lycopersicum | MT673878.1 | MT673879.1 | 2019 |
| ToCV-BJ | China | Solanum lycopersicum | KC887998.1 | KC887999.1 | 2007 |
| ToCV-PAP-JJ6 | Republic of Korea, Jinju | Capsicum annuum | OR865222.1 | OR865223.1 | 2023-05-01 |
| ToCV-SH-SIA | China | not applicable | MW490611.1 | MW490612.1 | 2019-11-01 |
| ToCV-SH-SIPPE | China | not applicable | MW490609.1 | MW490610.1 | 2019-11-01 |
| ToCV-SH-SNU | China | not applicable | MW490607.1 | MW490608.1 | 2019-11-01 |
| WSE | Egypt, Cairo, ARC | Solanum lycopersicum | KY618796.1 | KY618797.1 | 2022-05-18 |
| XS | Taiwan, Taichung, Xinshe | Solanum lycopersicum | KP114526.1 | KP114528.1 | 2011-12-19 |
| YG | Republic of Korea | Solanum lycopersicum | LC711108.1 | LC711109.1 | 2020 |
| Mie_1806 | Japan | Solanum lycopersicum | LC528262.1 | LC528263.1 | 2018 |
| Florida1 | USA, Florida | Solanum lycopersicum | NC_007340.1 | NC_007341.1 | 1989 |
| Florida2 | USA, Florida | Solanum lycopersicum | AY903447.1 | AY903448.1 | 1989 |
| FERA_160205 | Slovenia | Solanum lycopersicum | KY810786.1 | KY810787.1 | 2016 |
| TCG | Japan, Tochigi | Solanum lycopersicum | LC711106.1 | LC711107.1 | 2008 |
| KGS RNA | Japan, Kagoshima | Solanum lycopersicum | LC791014.1 | LC794715.1 | 2022-05-18 |
| Peptide ID | 9-mer Peptide | Target Protein |
|---|---|---|
| ToCV_EPI1 | YMLESVKNV | YP_293694.1 (1a polyprotein) |
| ToCV_EPI2 | YTIDGILEL | YP_293699.1 (HSP70) |
| ToCV_EPI3 | KITDLNVSV | YP_293699.1 (HSP70) |
| ToCV_EPI4 | TMNSVVQYV | YP_293704.1 (Minor coat protein) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Kim, J. Discovery of Targetable Epitopes in Tomato Chlorosis Virus Through Comparative Genomics and Structural Modeling. Sci 2025, 7, 88. https://doi.org/10.3390/sci7030088
Choi BY, Kim J. Discovery of Targetable Epitopes in Tomato Chlorosis Virus Through Comparative Genomics and Structural Modeling. Sci. 2025; 7(3):88. https://doi.org/10.3390/sci7030088
Chicago/Turabian StyleChoi, Bae Young, and Jaewook Kim. 2025. "Discovery of Targetable Epitopes in Tomato Chlorosis Virus Through Comparative Genomics and Structural Modeling" Sci 7, no. 3: 88. https://doi.org/10.3390/sci7030088
APA StyleChoi, B. Y., & Kim, J. (2025). Discovery of Targetable Epitopes in Tomato Chlorosis Virus Through Comparative Genomics and Structural Modeling. Sci, 7(3), 88. https://doi.org/10.3390/sci7030088