Thermodynamic Stability, Thermoelectric, Elastic and Electronic Structure Properties of ScMN2-Type (M = V, Nb, Ta) Phases Studied by ab initio Calculations


Abstract
:1. Introduction
2. Computational Details
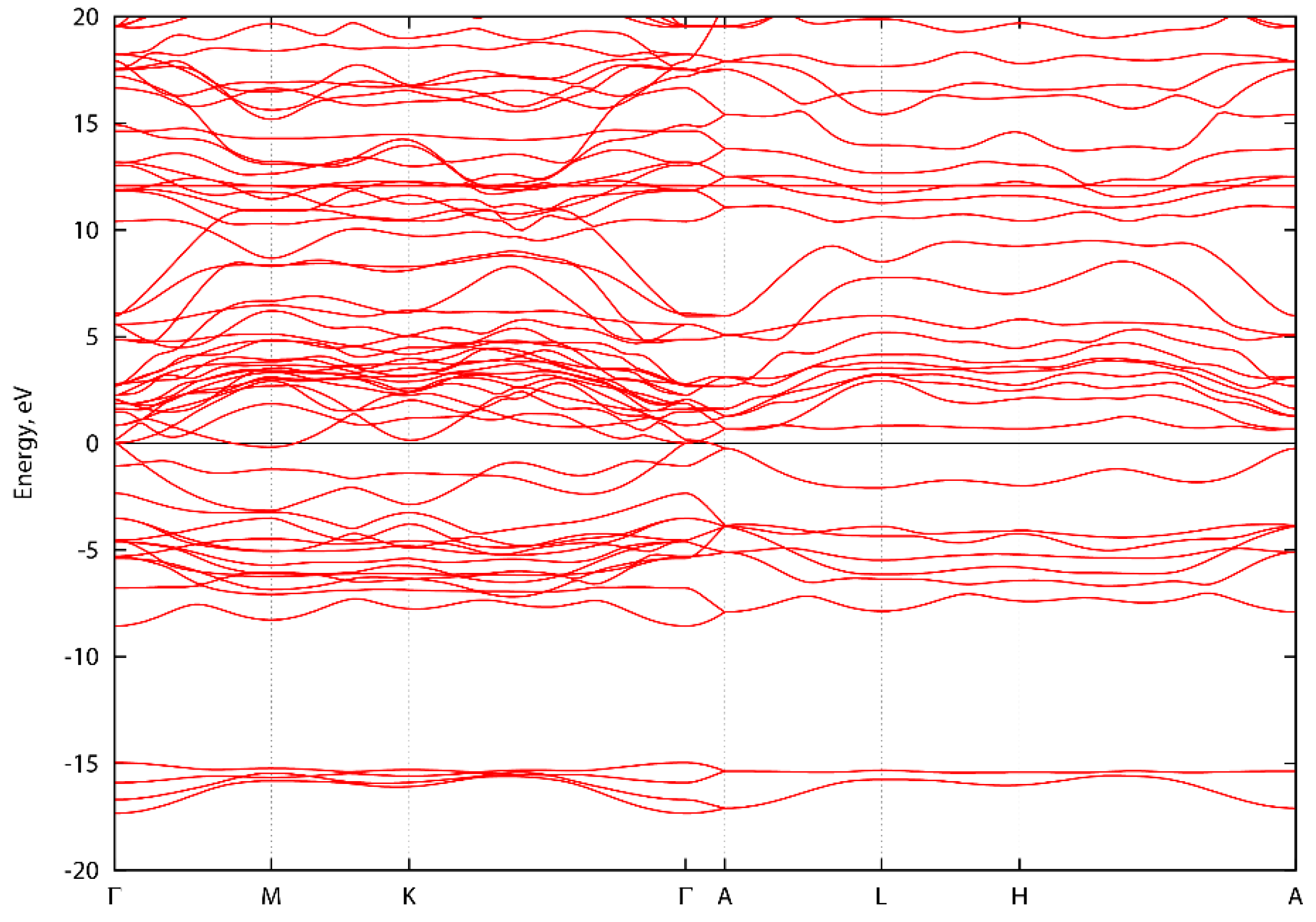
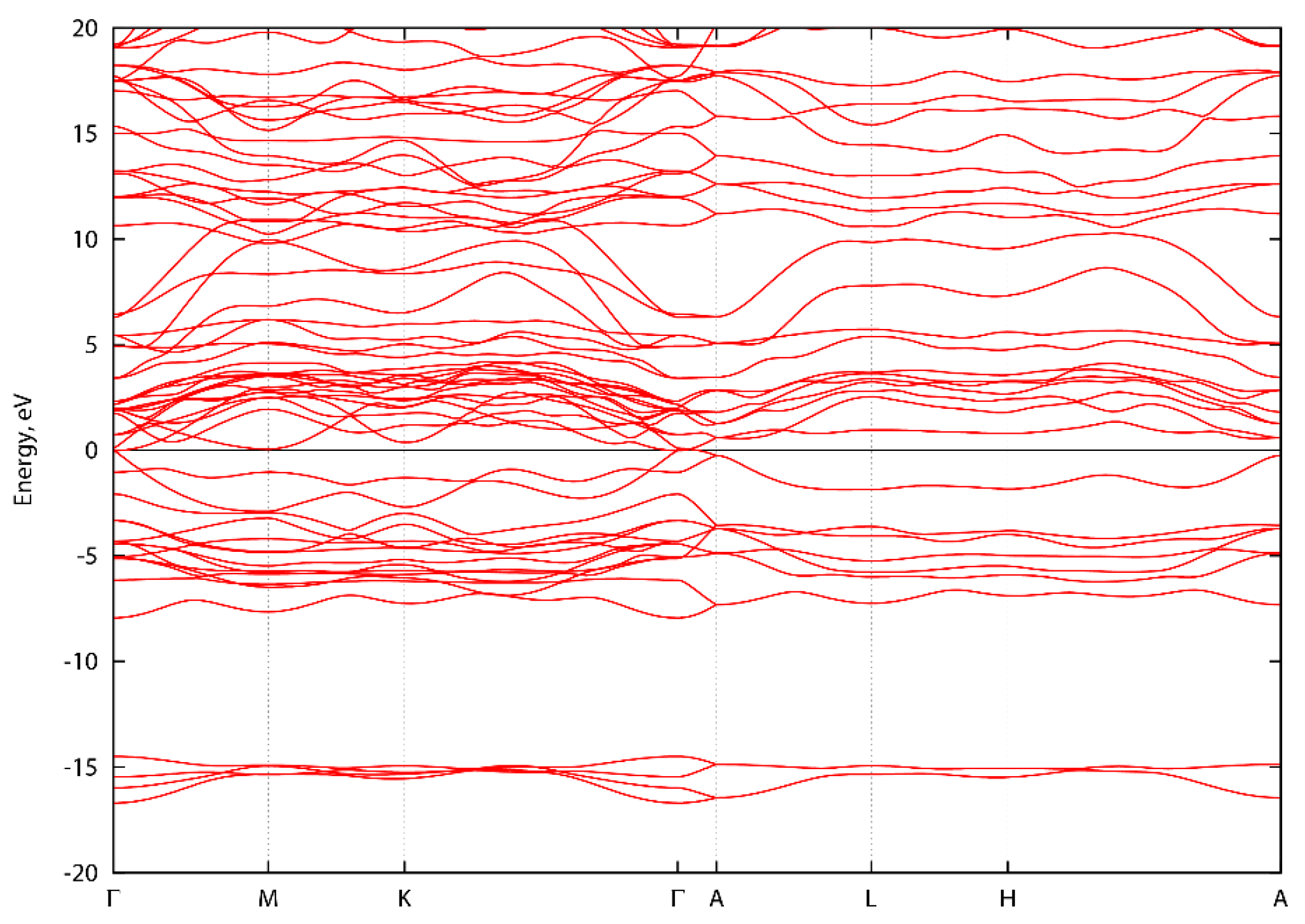
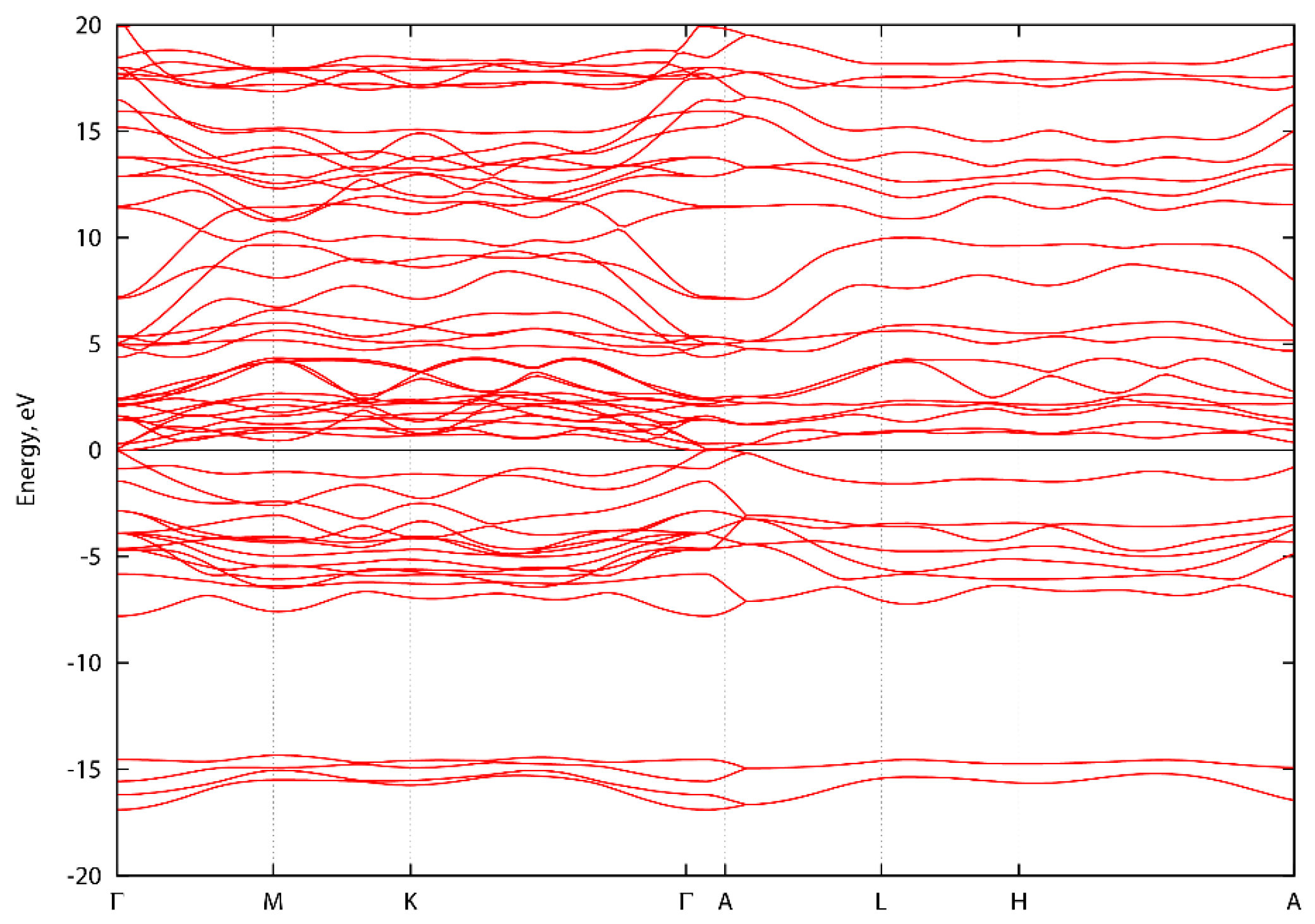
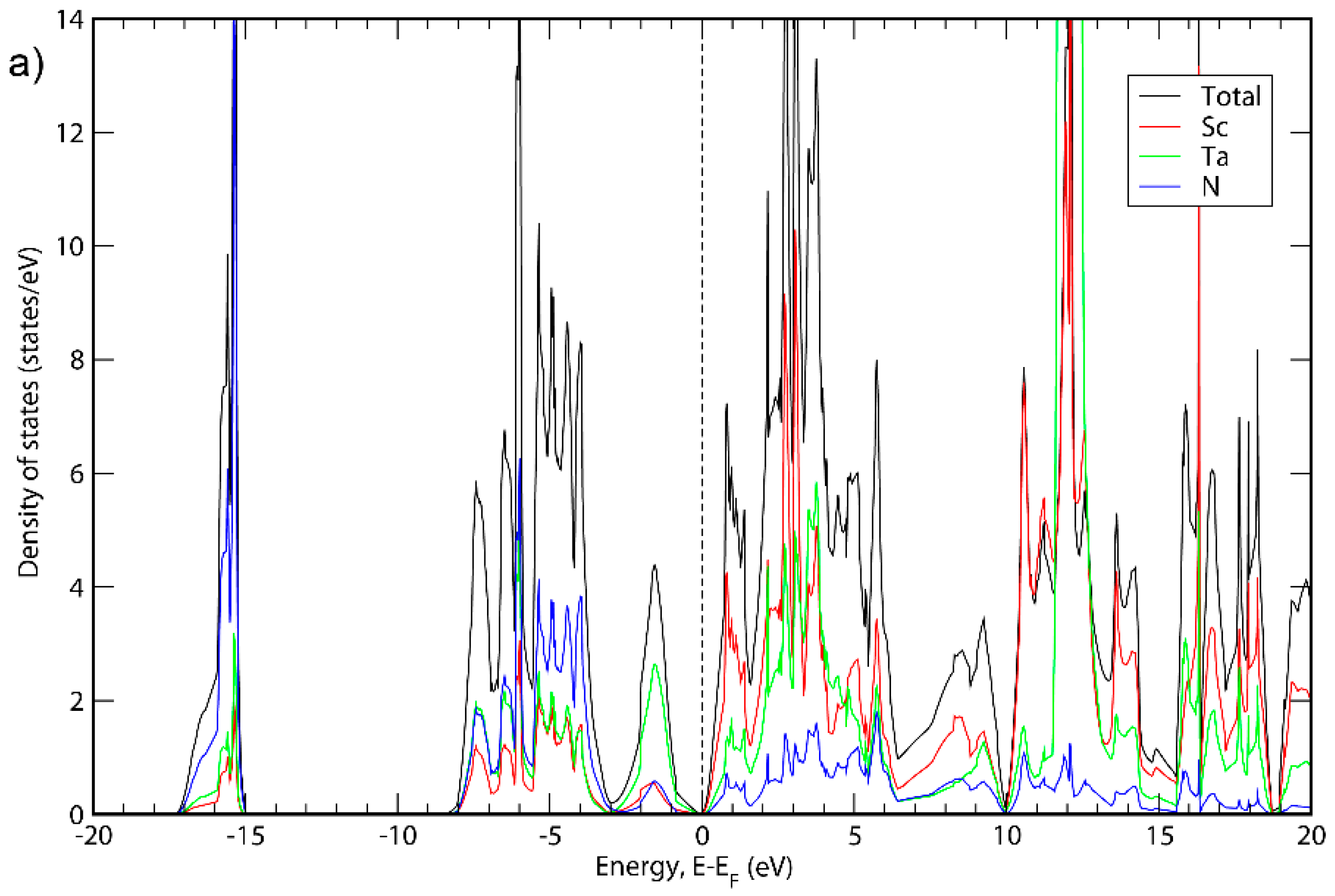
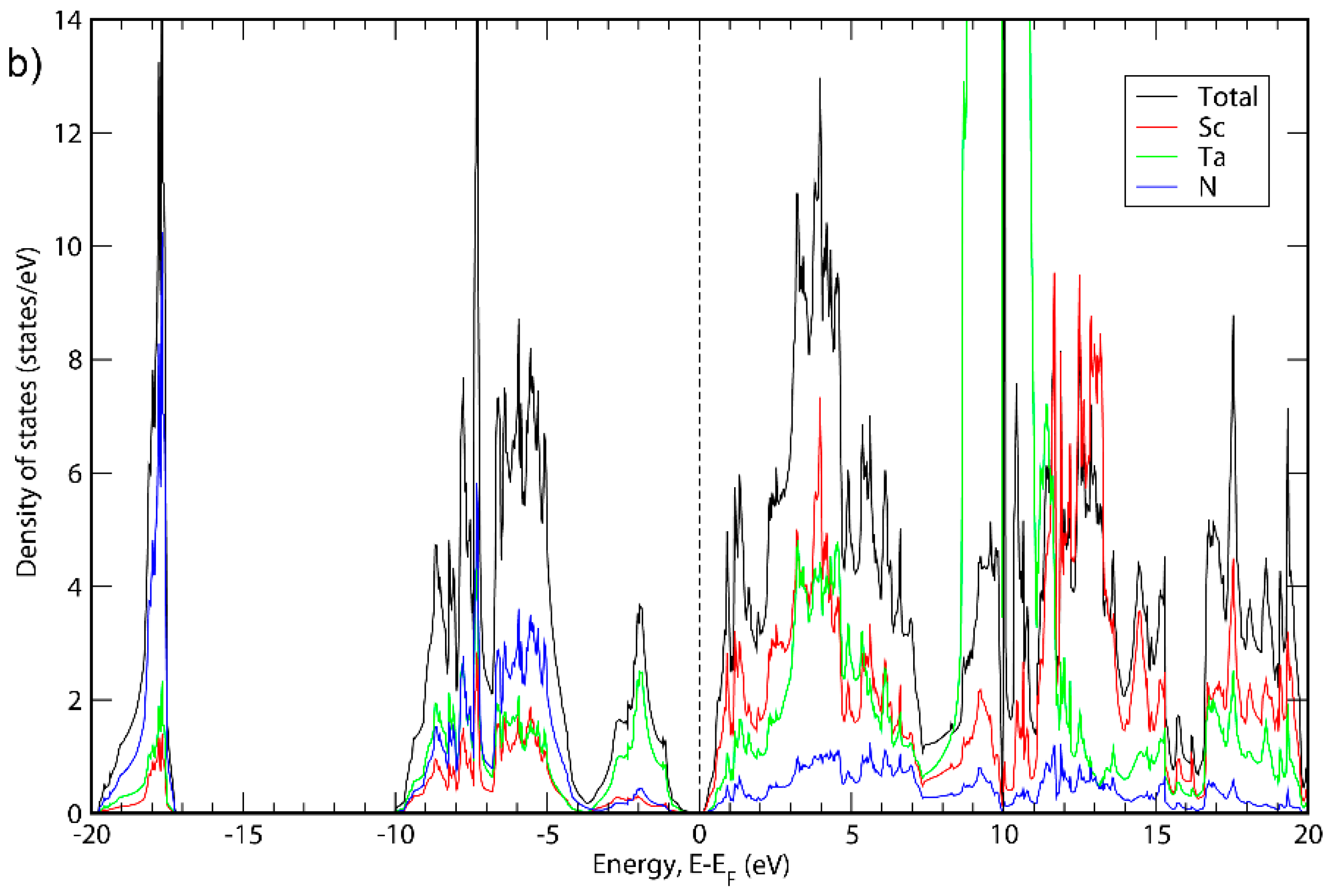
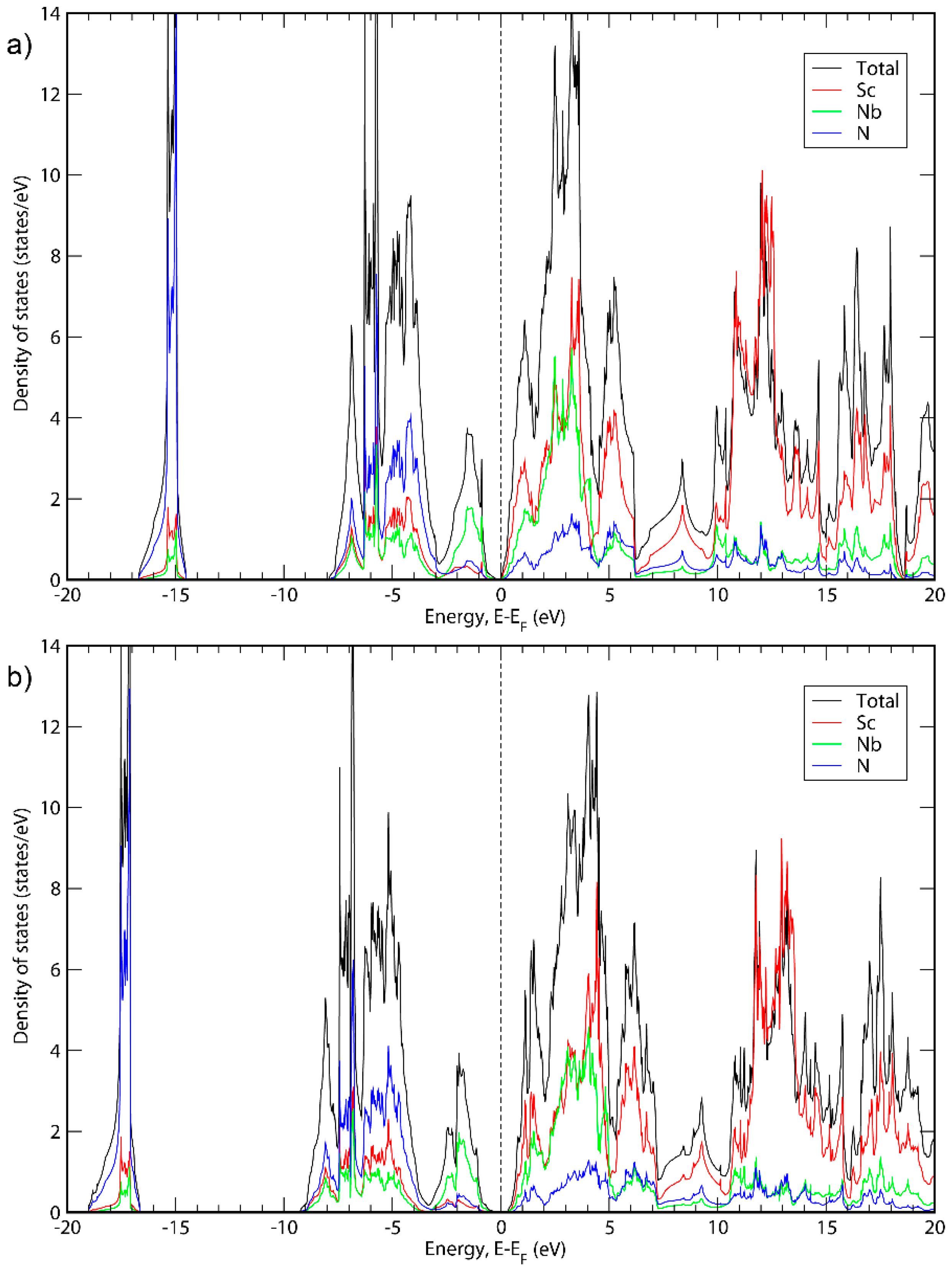
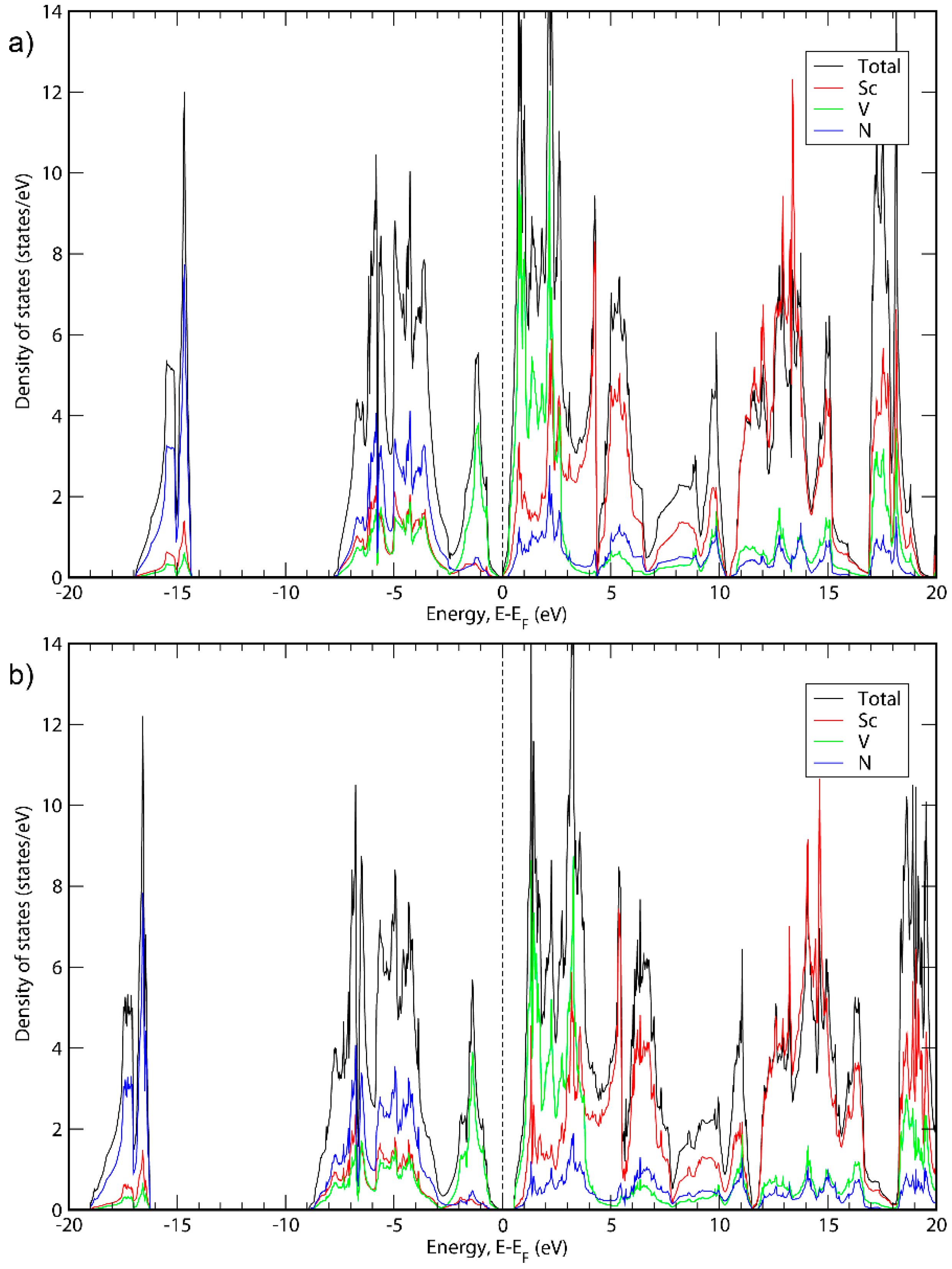
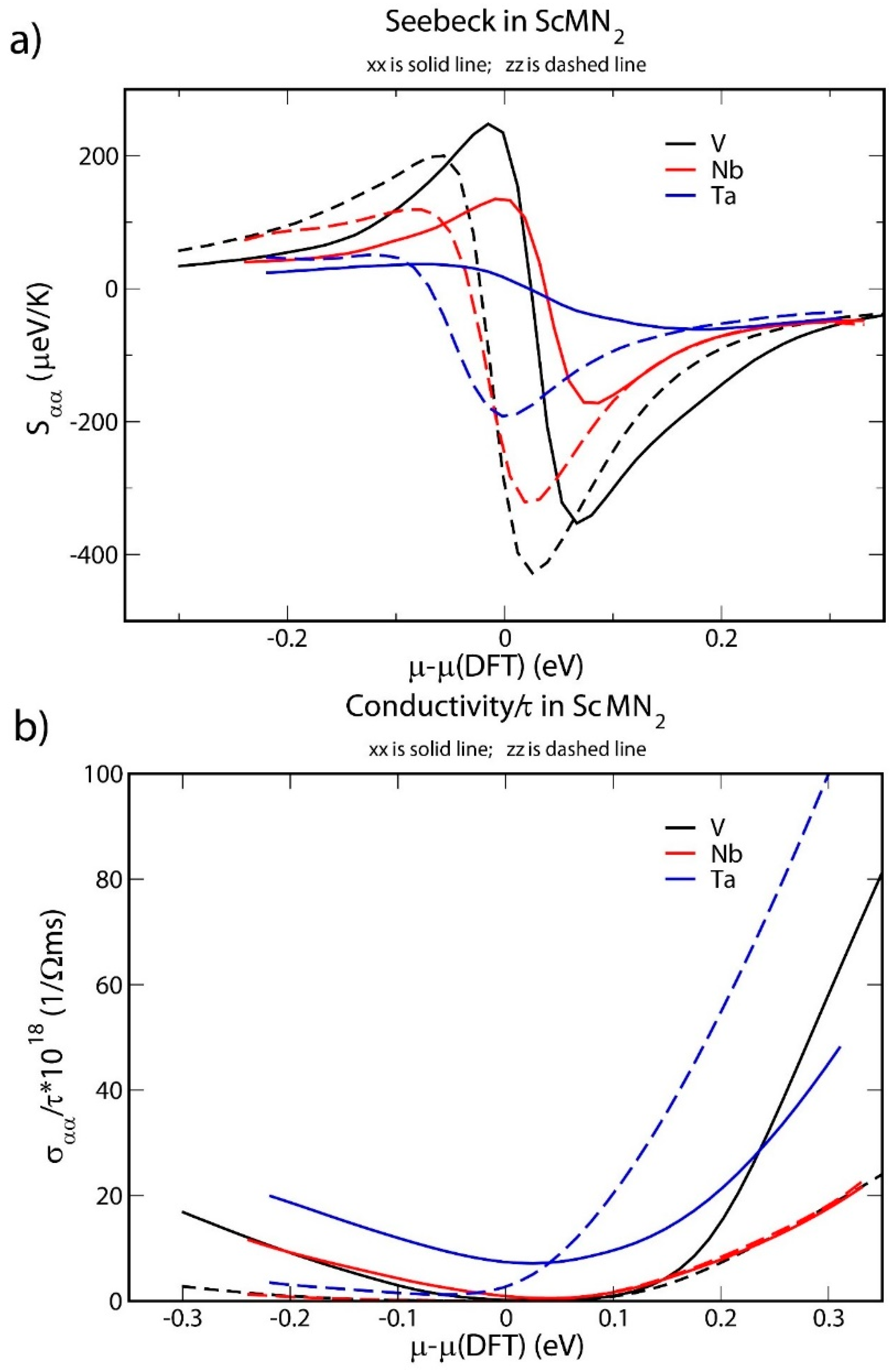
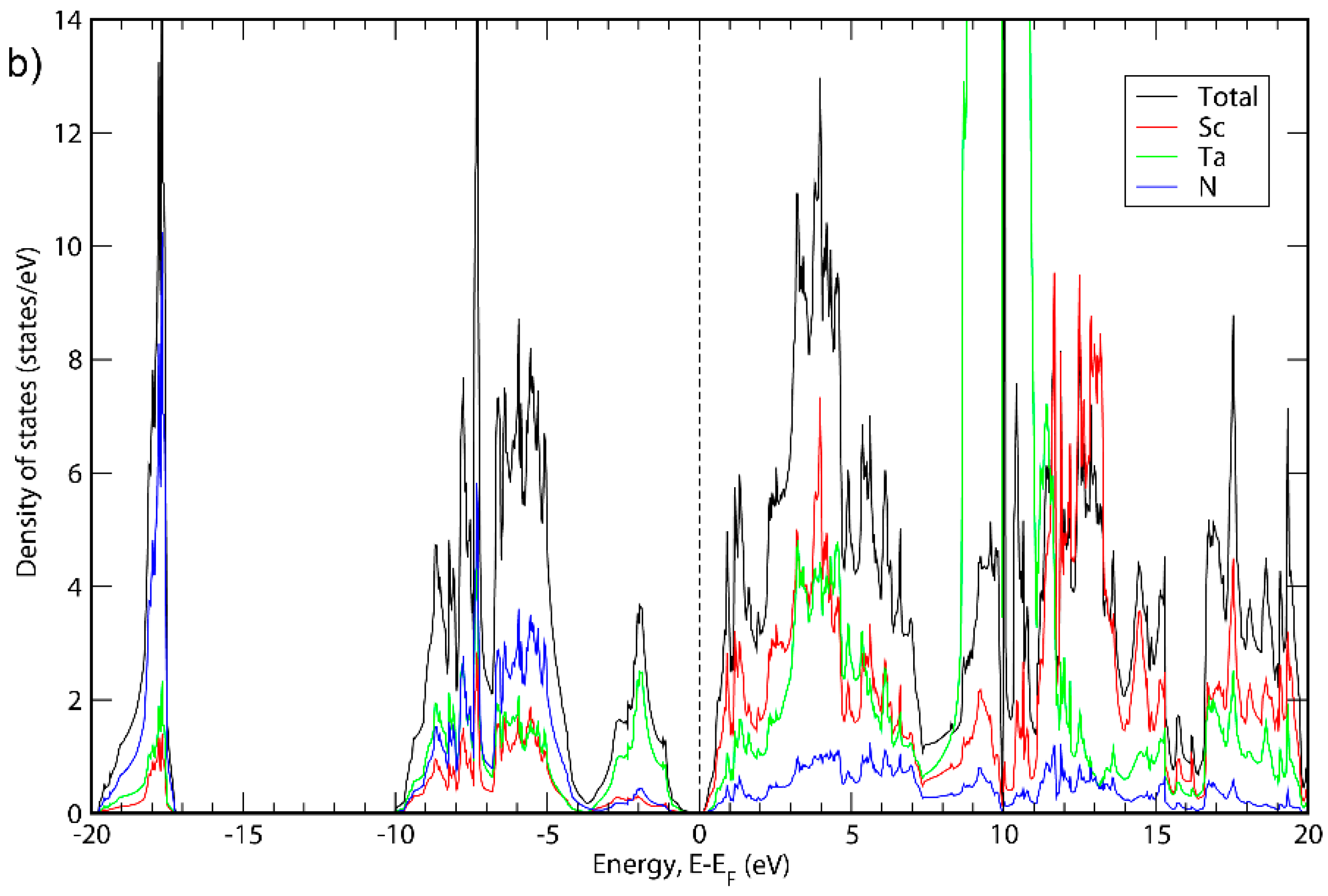
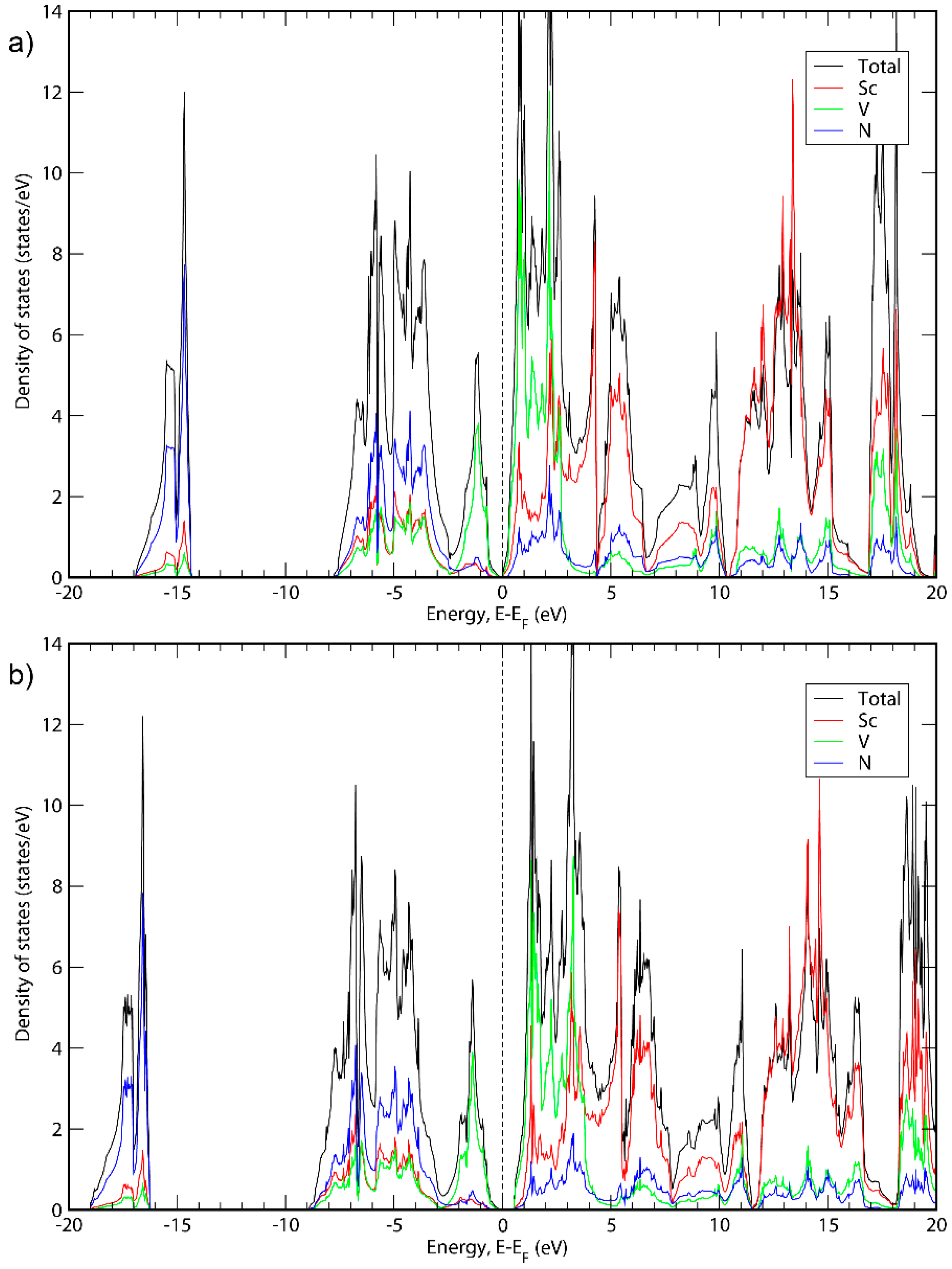
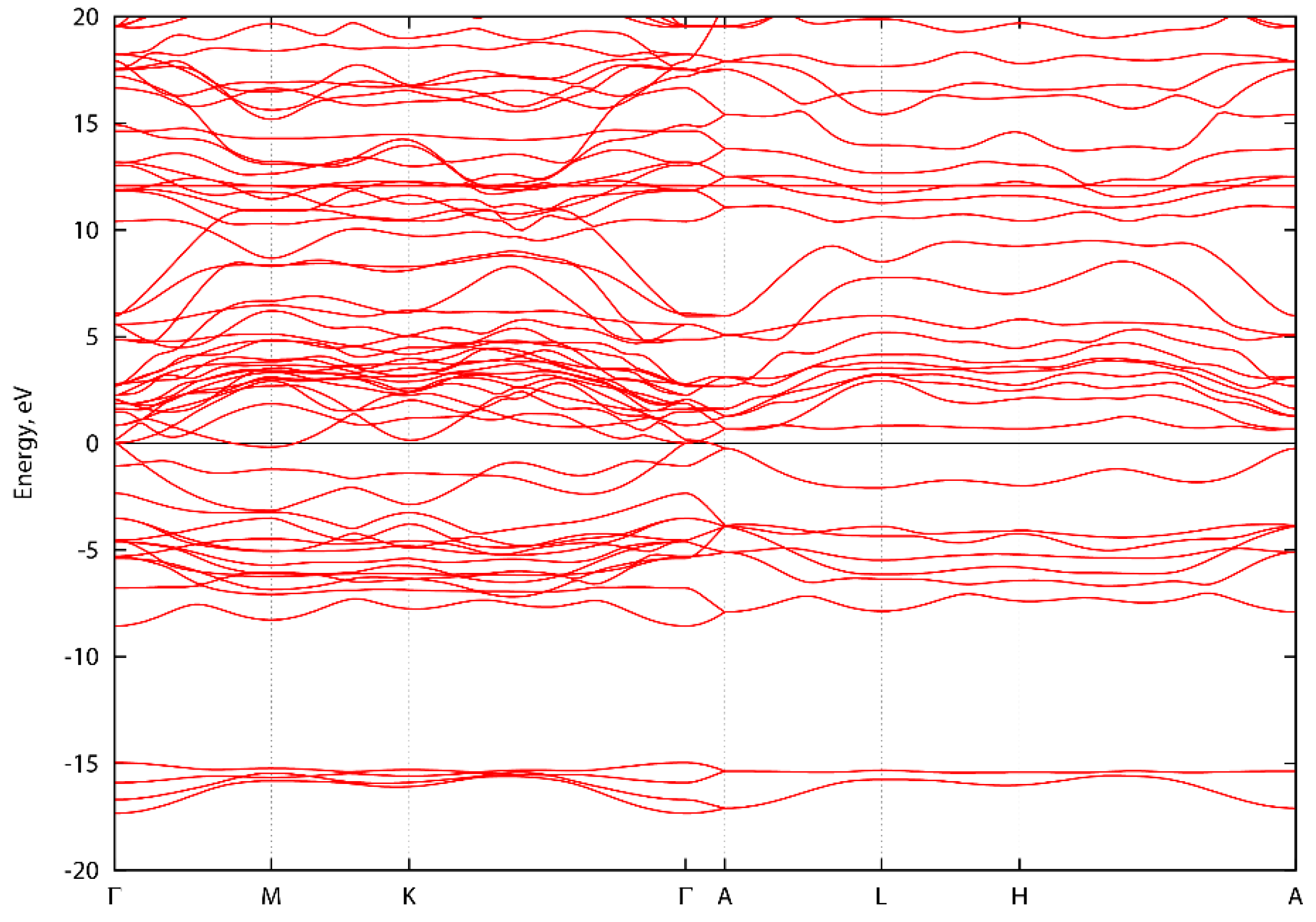
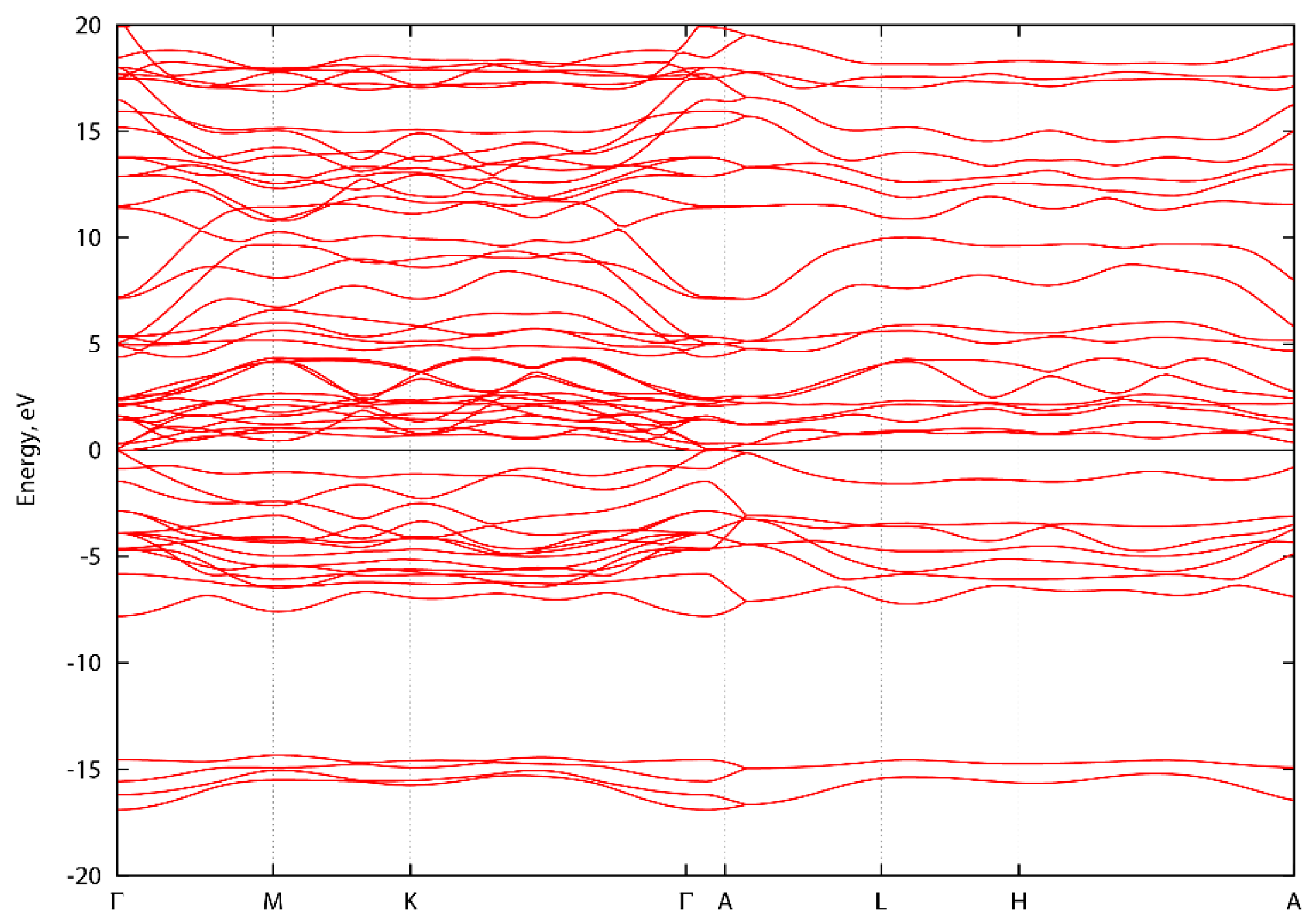
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Eklund, P.; Beckers, M.; Jansson, U.; Högberg, H.; Hultman, L. The Mn+1AXn phases: Materials science and thin-film processing. Thin Solid Films 2010, 518, 1851–1878. [Google Scholar] [CrossRef]
- Eklund, P.; Rosén, J.; Persson, P.O.A. Layered ternary Mn+1AXn phases and their 2D derivative MXene: An overview from a thin-film perspective. J. Phys. D 2017, 50, 113001. [Google Scholar] [CrossRef]
- Magnuson, M.; Mettesini, M. Chemical Bonding and Electronic-Structure in MAX phases as Viewed by X-ray Spectroscopy and Density Functional Theory. Thin Solid Films 2017, 621, 108–130. [Google Scholar] [CrossRef]
- Radovic, M.; Barsoum, M.W. MAX phases: Bridging the gap between metals and ceramics. Am. Ceram. Soc. Bull. 2013, 92, 20–27. [Google Scholar]
- Sun, Z.M. Progress in research and development on MAX phases: a family of layered ternary compounds. Int. Mater. Rev. 2013, 56, 143–166. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhou, Y.C. Layered Materials and Electrically Conductive Ti2AlC and Ti3AlC2 Ceramics: A Review. J. Mater. Sci. Technol. 2010, 26, 385–416. [Google Scholar] [CrossRef]
- Barsoum, M.W. The MN+1AXN Phases: A New Class of Solids; Thermodynamically Stable Nanolaminates. Prog. Solid State 2000, 28, 201–281. [Google Scholar] [CrossRef]
- Barsoum, M.W. The MAX Phases: Unique New Carbide and Nitride Materials. Am. Sci. 2001, 89, 334–343. [Google Scholar] [CrossRef]
- Lengauer, W. The Crystal Structure of ScNbN1-x and Comparisons with Related Nitride and Carbide Structures. J. Solid State Chem. 1989, 82, 186–191. [Google Scholar] [CrossRef]
- Lengauer, W.; Ettmayer, P. THE CRYSTAL STRUCTURE OF ScTaN1-x. J. Less Common Met. 1988, 141, 157–162. [Google Scholar] [CrossRef]
- Niewa, R.; Zherebtsov, D.A.; Schnelle, W.; Wagner, F.R. Metal-Metal Bonding in ScTaN2. A New Compound in the System ScN-TaN. Inorg. Chem. 2004, 43, 6188–6194. [Google Scholar] [PubMed]
- Kerdsongpanya, S.; Alling, B.; Eklund, P. Phase stability of ScN-based solid solutions for thermoelectric applications from first-principles calculations. J. Appl. Phys. 2013, 114, 073512. [Google Scholar] [CrossRef]
- Burmistrova, P.V.; Maassen, J.; Favaloro, T.; Saha, B.; Salamat, S.; Rui Koh, Y.; Lundstrom, M.S.; Shakouri, A.; Sands, T.D. Thermoelectric properties of epitaxial ScN films deposited by reactive magnetron sputtering onto MgO(001) substrates. Appl. Phys. 2013, 113, 153704. [Google Scholar] [CrossRef]
- Burmistrova, P.V.; Zakharov, D.N.; Favaloro, T.; Mohammed, A.; Stach, E.A.; Shakouri, A.; Sands, T.D. Effect of deposition pressure on the microstructure and thermoelectric properties of epitaxial ScN(001) thin films sputtered onto Mgo(001) substrates. J. Mater. Res. 2015, 30, 626–634. [Google Scholar] [CrossRef]
- Gall, D.; Petrov, I.; Hellgren, N.; Hultman, L.; Sundgren, J.E.; Greene, J.E. Growth of poly- and single-crystal ScN on MgO(001): Role of low-energy N+2 irradiation in determining texture, microstructure evolution, and mechanical properties. J. Appl. Phys. 1998, 84, 6034–6041. [Google Scholar] [CrossRef]
- Kerdsongpanya, S.; Alling, B.; Eklund, P. Effect of point defects on the electronic density of states of ScN studied by first-principles calculations and implications for thermoelectric properties. Phys. Rev. B 2012, 86, 195140. [Google Scholar] [CrossRef]
- Le Febvrier, A.; Tureson, N.; Stilkerich, N.; Greczynski, G.; Eklund, P. Effect of impurities on morphology, growth mode, and thermoelectric properties of (111) and (001) epitaxial-like ScN films. J. Phys. D Appl. Phys. 2019, 52, 035302. [Google Scholar] [CrossRef]
- Kerdsongpanya, S.; Hellman, O.; Sun, B.; Koh, Y.K.; Lu, J.; Van Nong, N.; Simak, S.I.; Alling, B.; Eklund, P. Phonon thermal conductivity of scandium nitride for thermoelectrics from first-principles calculations and thin-film growth. Phys. Rev. B 2017, 96, 195417. [Google Scholar] [CrossRef]
- Kerdsongpanya, S.; Sun, B.; Eriksson, F.; Jensen, J.; Lu, J.; Koh, Y.K.; Nong, N.V.; Balke, B.; Alling, B.; Eklund, P. Experimental and theoretical investigation of Cr1-xScxN solid solutions for thermoelectrics. J. Appl. Phys. 2016, 120, 215103. [Google Scholar] [CrossRef]
- Saha, B.; Garbrecht, M.; Perez-Taborda, J.A.; Fawey, M.H.; Koh, Y.R.; Shakouri, A.; Martin-Gonzalez, M.; Hultman, L.; Sands, T.D. Compensation of native donor doping in ScN: Carrier concentration control and p-type ScN. Appl. Phys. Lett. 2017, 110, 252104. [Google Scholar] [CrossRef]
- Tureson, N.; Van Nong, N.; Fournier, D.; Singh, N.; Acharya, S.; Schmidt, S.; Belliard, L.; Soni, A.; Eklund, P. Reduction of the thermal conductivity of the thermoelectric material ScN by Nb alloying. J. Appl. Phys. 2017, 122, 025116. [Google Scholar] [CrossRef]
- Rawat, V.; Koh, Y.K.; Cahill, D.G.; Sands, T.D. Thermal conductivity of (Zr,W)N/ScN metal/semiconductor multilayers and superlattices. J. Appl. Phys. 2009, 105, 024909. [Google Scholar] [CrossRef]
- Saha, B.; Koh, Y.R.; Comparan, J.; Sadasivam, S.; Schroeder, J.L.; Garbrecht, M.; Mohammed, A.; Birch, J.; Fisher, T.; Shakouri, A.; et al. Cross-plane thermal conductivity of (Ti;W)N/(Al,Sc)N metal. Phys. Rev. B 2016, 93, 045311. [Google Scholar] [CrossRef]
- Schroeder, J.L.; Saha, B.; Garbrecht, M.; Schell, N.; Sands, T.D.; Birch, J. Thermal stability of epitaxial cubic-TiN/(Al,Sc)N metal/semiconductor superlattices. J. Mater. Sci. 2015, 50, 3200. [Google Scholar] [CrossRef]
- Zebarjadi, M.; Bian, Z.; Singh, R.; Shakouri, A.; Wortman, R.; Rawat, V.; Sands, T. Thermoelectric Transport in a ZrN/ScN Superlattice. J. Elctron Mater. 2009, 38, 960–963. [Google Scholar] [CrossRef]
- Gharavi, M.A.; Kerdsongpanya, S.; Schmidt, S.; Eriksson, F.; Nong, N.V.; Lu, J.; Balke, B.; Fournier, D.; Belliard, L.; le Febvrier, A.; et al. Microstructure and thermoelectric properties of CrN and CrN/Cr2N thin films. J. Phys. D 2018, 51, 355302. [Google Scholar] [CrossRef]
- Ravi, C. First-principles study of ground-state properties and phase stability of vanadium nitrides. CALPHAD 2009, 33, 469–477. [Google Scholar] [CrossRef]
- Grumski, M.; Dholabhai, P.P.; Adams, J.B. Ab initio study of the stable phases of 1:1 tantalum nitride. Acta Mater. 2013, 61, 3799–3807. [Google Scholar] [CrossRef]
- Mei, A.B.; Hellman, O.; Wireklint, N.; Schlepuetz, C.M.; Sangiovanmi, D.; Alling, B.; Rockett, A.; Hultman, L.; Petrov, I.; Greene, J.E. Dynamic and structural stability of cubic vanadium nitride. Phys. Rev. B 2015, 91, 054101. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 54, 11169. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamis simulation of the liquid-metal-amorphous-semiconductor transition in germaniium. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, F.; Ernzerhof, M. Generalized Gradient Approxiamtion Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic data for strained materials from ab initio calculations of stress. Phys. Rev. B 2002, 65, 104104. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.; Kvasnicka, D.; Luitz, J. WIEN2k, An augmented Plane Wave + Local Orbitals for Calculating Crystal Properteis; Techn. Universität Wien: Vienna, Austria, 2001. [Google Scholar]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Thore, A.; Dahlqvist, M.; Alling, B.; Rosén, J. Temperature dependent phase stability of nanolaminated ternaries from first-principles calculations. Comput. Mater. Sci. 2014, 91, 251–257. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Haas, P.; Tran, F.; Blaha, P. Calculation of the lattice constant of solids with semilocal functionals. Phys. Rev. B 2009, 79, 085109. [Google Scholar] [CrossRef]
- Schimka, L.; Harl, J.; Kresse, G. Improved hybrid functional for solids: the HSEsol functional. J. Chem. Phys. 2011, 134, 024116. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Fließgrenze von Mischkristrallen auf der Grund der Plastizitätsbedingung für Einkristalle. Z. Angew. Math. Mech. 1929, 9, 55. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. Ser. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Barsoum, M.W. Mechanical properties of the MAX Phases. Annu. Rev. Mater. Res. 2011, 41, 195–227. [Google Scholar] [CrossRef]
- Bagayoko, D. Understanding density functional theory (DFT) and completing it in practice. AIP Adv. 2014, 4, 127104. [Google Scholar] [CrossRef]
- Kerdsongpanya, S.; Nong, N.V.; Pryds, N.; Žukauskaitė, A.; Jensen, J.; Birch, J.; Lu, J.; Hultman, L.; Wingqvist, G.; Eklund, P. Anomalously high thermoelectric power factor in epitaxial ScN thin films. Appl. Phys. Lett. 2011, 99, 132113. [Google Scholar] [CrossRef]
- Lambrecht, W.R.L. Electronic structure and optical spectra of the semimetal ScAs and of the indirect-band-gap semiconductors ScN and GdN. Phys. Rev. B 2000, 62, 13538. [Google Scholar] [CrossRef]
- Tran, F.; Blaha, P. Accurate Band Gaps of Semiconductors and Insulators with a Semilocal Exchange-Correlation Potential. Phys. Rev. Lett. 2009, 102, 226401. [Google Scholar] [CrossRef]
- Chaput, L.; Hug, G.; Pécher, P.; Scherrer, H. Thermopower of the 312 MAX phases Ti3 Si C2, Ti3 Ge C2 and Ti3 Al C2. Phys. Rev. B 2007, 75, 035107. [Google Scholar] [CrossRef]
- Chaput, L.; Hug, G.; Pécher, P.; Scherrer, H. Anisotropy and thermopower in Ti3SiC2. Phys. Rev. B 2005, 71, 121104. [Google Scholar] [CrossRef]
- Yoo, H.-I.; Barsoum, M.W.; El-Raghy, T. Ti3SiC2 has negligible thermopower. Nature 2000, 407, 581–582. [Google Scholar] [CrossRef]
- Saha, B.; Perez-Taborda, J.A.; Bahk, J.-H.; Koh, Y.R.; Shakouri, A.; Martin-Gonzalez, M.; Sands, T.D. Temperature-dependent thermal and thermoelectric properties of n-type and p-type Sc1-xMgxN. Phys. Rev. B 2018, 97, 085301. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ScTaN2 | ||||
| a (Å) | 3.0534 | |||
| c (Å) | 10.5685 | |||
| Atom | Site | x | y | z |
| Sc | 2a | 0 | 0 | 0 |
| Ta | 2d | 1/3 | 2/3 | 3/4 |
| N | 4f | 1/3 | 2/3 | 0.1231 |
| ScNbN2 | ||||
| a (Å) | 3.0633 | |||
| c (Å) | 10.5702 | |||
| Atom | Site | x | y | z |
| Sc | 2a | 0 | 0 | 0 |
| Nb | 2d | 1/3 | 2/3 | 3/4 |
| N | 4f | 1/3 | 2/3 | 0.1250 |
| Parameter | ScTaN2 | ScNbN2 | ScVN2 |
|---|---|---|---|
| [eV] | −1.07 | −0.84 | −0.22 |
| C11 [GPa] | 551 | 522 | 480 |
| C12 [GPa] | 158 | 152 | 145 |
| C13 [GPa] | 143 | 130 | 121 |
| C33 [GPa] | 552 | 546 | 572 |
| C44 [GPa] | 196 | 189 | 167 |
| a [Å] | 3.0791 | 3.0824 | 2.9774 |
| c [Å] | 10.6254 | 10.6060 | 10.2591 |
| z | 0.1238 | 0.1237 | 0.1313 |
| Parameter | ScTaN2 | ScNbN2 | ScVN2 |
|---|---|---|---|
| GV [GPa] | 197 | 190 | 179 |
| BV [GPa] | 283 | 268 | 256 |
| EV [GPa] | 479 | 460 | 436 |
| GR [GPa] | 197 | 189 | 175 |
| BR [GPa] | 283 | 268 | 255 |
| ER [GPa] | 479 | 460 | 432 |
| GR/BR | 0.70 | 0.71 | 0.70 |
| GV/BV | 0.70 | 0.71 | 0.70 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilemalm, R.; Pourovskii, L.; Mosyagin, I.; Simak, S.; Eklund, P. Thermodynamic Stability, Thermoelectric, Elastic and Electronic Structure Properties of ScMN2-Type (M = V, Nb, Ta) Phases Studied by ab initio Calculations. Condens. Matter 2019, 4, 36. https://doi.org/10.3390/condmat4020036
Pilemalm R, Pourovskii L, Mosyagin I, Simak S, Eklund P. Thermodynamic Stability, Thermoelectric, Elastic and Electronic Structure Properties of ScMN2-Type (M = V, Nb, Ta) Phases Studied by ab initio Calculations. Condensed Matter. 2019; 4(2):36. https://doi.org/10.3390/condmat4020036
Chicago/Turabian StylePilemalm, Robert, Leonid Pourovskii, Igor Mosyagin, Sergei Simak, and Per Eklund. 2019. "Thermodynamic Stability, Thermoelectric, Elastic and Electronic Structure Properties of ScMN2-Type (M = V, Nb, Ta) Phases Studied by ab initio Calculations" Condensed Matter 4, no. 2: 36. https://doi.org/10.3390/condmat4020036