1. Introduction
Silver nanoparticles (AgNPs) are used in a wide variety of applications and are one of the most studied nanoparticles (NPs) due to their antimicrobial capabilities and their applications in different industries as textile or wastewater treatment [
1]. Their biomedical applications have been widely reviewed, highlighting both the advantages and disadvantages of their use and detailing their antibacterial, antifungal [
2], antiviral, anti-inflammatory, and antiangiogenic (i.e., inhibition of new blood vessel formation) properties [
3]. Given their broad spectrum of applications and potential uses, different methodologies have been proposed for the synthesis of AgNPs, such as chemical reduction and biological reduction or physical methods such as laser irradiation [
4,
5,
6]. Depending on the synthesis method employed, the resulting AgNPs possess a variety of shapes and size distributions, which in turn influence their optical properties and the ability to interact with specific molecule [
7,
8].
One strategy to produce AgNPs is the chemical reduction synthesis, which precisely controls nanoparticle size and shape, while another strategy known as biological or green synthesis generates amorphous nanoparticles with nanoparticle surface functionalities that are useful for certain applications [
9,
10,
11]. This biological strategy has resulted in a variate set of morphologies (cubical, triangular, oval, pebble-like, or circular in shape) in a single or aggregated pattern [
12,
13] with a diameter size >30 nm [
14]. The use of ultrasonic energy in green AgNP synthesis has favored the formation of AgNPs with a smaller size and an improved monodispersity [
12,
13]. The use of ultrasonic energy and plant extracts for AgNP synthesis has proved to produce smaller AgNPs (diameter < 10 nm) with spherical morphologies [
14] than magnetic stirring (±40 nm;where ultrasound has been used to assist synthesis in different plant extracts [
2].
Size and morphology are essential for applications in plasmonic sensors, where sensitivity and specificity of detection are critical [
15]. Different challenges arise in the research and development of specific NPs and their applicability as biosensors, as follows: the stability of nanoparticles in the medium; the aggregation over time; the control of the growth of the nanoparticle crystals; the morphology; the size; and the size distribution [
16,
17]. showed that AgNPs modified by high-powered Light-Emitting Diode (LED) energy produce diverse morphologies that present different catalytic activities towards methylene blue (MB) degradation. Spherical AgNPs possessed a more negative z potential (mV); meanwhile, nanoparticles with cubic morphology presented a less negative potential. Also, the current response was found to be inversely proportional to the size of the nanostructure, consistent with the smaller surface area of AgNPs.
The monodispersity and stability of AgNPs are variables to consider over time because these characteristics are of great relevance in practical applications where consistency and reliability are required [
18] found a great stability of AgNPs reduced with citric acid and malic acid at pH > 7.0, where the stability of rods and quasi-spherical morphologies after 7 weeks of synthesis was found according to the SPR signal and SEM characterization. For AgNPs exposed to acidic and alkaline pH, the stability of silver ion aggregation into AgNPs was found to be pH dependent, showing NPs of large diameter (around 400 nm) at pH < 5.0 and a higher concentration of Ag
+ ions in solution. This contrasts with AgNPs synthetized at pH > 7.0, where the NP diameter was smaller and a lower Ag
+ ion concentration was found [
19]. In addition to the pH level, the concentration of salts in the reaction and the exposure to light also affect the aggregation of AgNPs [
20]. Another factor to contemplate is the use of high pressure on synthetized AgNPs. In the case of small AgNPs (5–10 nm) exposed to ultra-high pressure at the gigapascal (GPa) level, they undergo a structural distortion consistent with a rhombohedral distortion with increasing pressure.
Synthetized AgNPs can be used as electrochemical sensors or used as colorimetric detectors when surface plasmonic resonance (SPR) changes in the moment the nanoparticle specifically interacts with a specific compound [
21]. AgNP-based electrochemical sensors are based on the conversion of chemical reactions into measurable electrical signals, where kinetics of the redox reaction at the electrode surface influences the transduction efficiency [
22,
23,
24]. Also, AgNPs have been used as an antibacterial agent because their main action on bacteria is related to the interaction and modification of the external bacterial membrane [
25,
26], the accumulation of AgNPs on bacterial membranes allows the penetration of particles and the perturbation of membrane permeability. These effects are caused by the production of Ag+ ions from the oxidative dissolution of AgNPs, which is favored by smaller particles because the small particles dissolve faster, releasing more Ag
+. In Addition to nanoparticle size, cubic-shaped nanoparticles present better antibacterial activities than nanospheres and nanowires due to the closer contact of nanocubes to the bacterial surface, which has been explained by the higher reactivity of facet (100) in nanocubes than the reactivity of facet (111) in nanospheres [
27].
On the other hand, to increase specificity, the AgNPs can be further functionalized and bioconjugated to allow the specific detection of substances. This functionalization generates changes in the SPR and provides a quantitative signal that can be used to determine the presence and concentration of the detected substance [
28]. In the same way that the higher homogeneity of NPs is achieved, the higher sensitivity of AgNPs is reached due to a sharper and more defined SPR peak [
29]. In the case of capture probes like immunosensors, the AgNPs are bioconjugated to antibodies to enhance probe selectivity toward targeted analytes and to immobilize directly to the electrode surface to enhance selectivity and further quantify the amount of analyte detected [
30,
31,
32]. Also, as the proportion of available atoms at the surface of a nanoparticle increases, the number of capping molecules does as well [
33].
Computational simulations correlated the surface functionalization area with the van der Waals energy and counterion interactions. Results demonstrate that the thiol molecule’s surface density saturation on gold nanoparticles (AuNPs) reaches less than 75% of coverage, if each sulfur atom of a thiol chain with chemical composition S-[CH
2]
2-CH
3 is covalently bonded to the surface gold atoms of AuNPs [
34,
35,
36,
37]. In the same way, at low thiol surface densities, these are arranged in a quasi-parallel orientation respecting the AuNP surface; however, at 50% of coverage, thiol chains are tightly packaged and are mostly loose from the surface. Finally, for larger AuNPs, the surface curvature is smaller, and the access of counterions near the thiol-modified AuNPs proves to be more difficult than smaller AuNPs. In addition to the number of surface atoms that are functionalized, the total number of functional groups in a functionalized nanoparticle determines the zeta potential and, in consequence, their colloidal stability, dispersibility, and hydrophobicity or hydrophilicity, as well as their size, size distribution, shape, and morphology [
38]. As mentioned previously, the surface functionalization of NPs can be covered below 75% because of the bulkiness of the functionalization compound and the steric effect caused by its chemical structure [
39]. Also, the type of chemical bond produced between the surface and the functionalization compound should be considered. For example, in the case of tightly covalent carbon chains attached to the NP surface, only steric effects affect the accessibility of the carbon chain [
40].
Because size is an important issue to consider if NPs are synthetized to be functionalized and bioconjugated for specific detection of molecules, the main objective of this work was to evaluate the effect of pH and pressure on the size and plasmonic characteristics of AgNPs by using ultrasound energy as a rapid and cost-effective synthesis method and to develop different methods that allow the production of AgNPs with a specific size.
Although several studies have explored the influence of pH and other parameters on the size, morphology, and stability of silver nanoparticles (AgNPs), the combined effect of pressure and pH on nanoparticle size remains insufficiently addressed. Research has shown that acidic and alkaline conditions significantly affect AgNP aggregation and ion release, while high-pressure environments can induce structural changes at the nanoscale. However, there is still a lack of systematic studies evaluating how pressure and pH interact to determine nanoparticle size and monodispersity, especially when using rapid and scalable green synthesis approaches. Conventional methods often result in broad size distributions or require longer synthesis times and post-processing steps. This study addresses these challenges by employing ultrasound-assisted synthesis—a method known for enhancing nucleation and reducing reaction times—to evaluate how varying pH and applied pressure conditions affect the formation and size of AgNPs. By doing so, the study offers a cost-effective and time-efficient strategy to control nanoparticle size with higher precision, which is crucial for the use of AgNPs in sensitive applications such as biosensors and antimicrobial systems.
3. Results
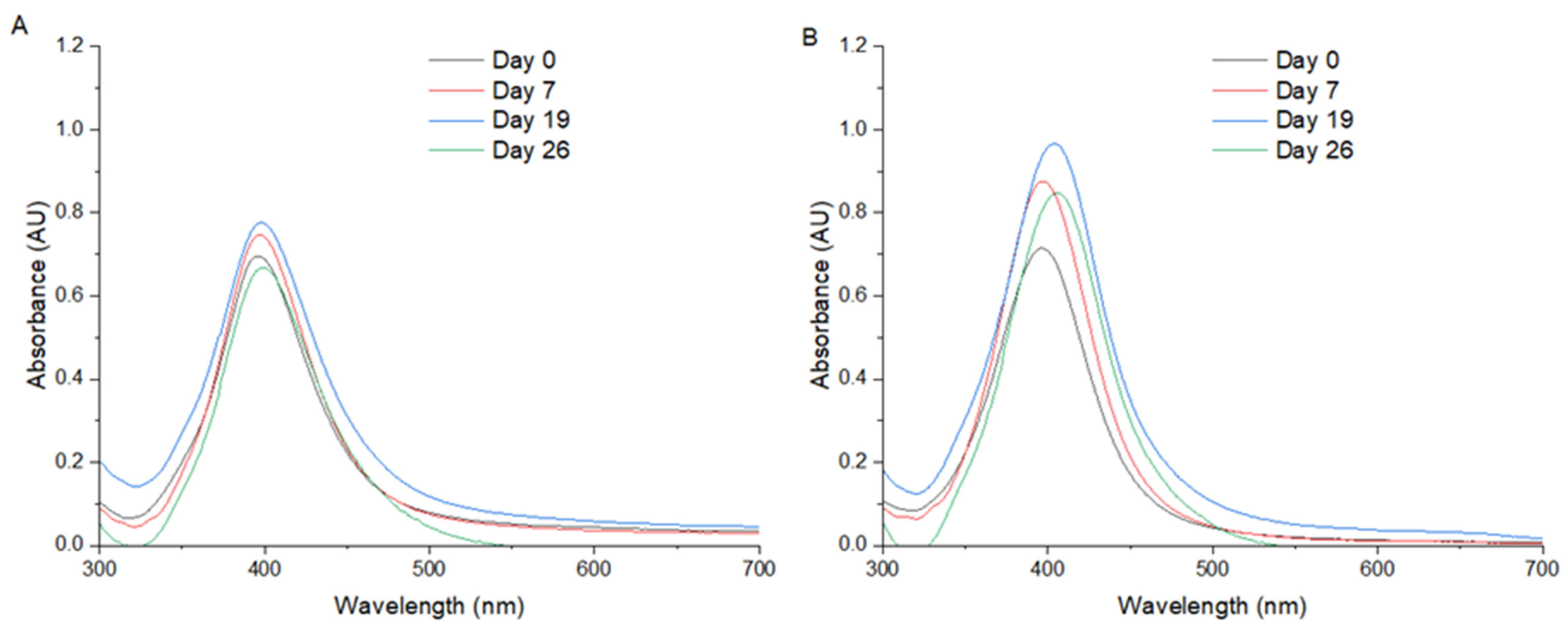
Figure 1A,B display the UV/Vis spectra of AgNPs synthesized via Method 1 and Method 2, respectively. In both cases, a distinct and symmetric SPR peak is observed at 396 nm, which is typically associated with the formation of well-dispersed, spherical silver nanoparticles with diameters in the range of 10–20 nm. The symmetry and narrowness of the SPR peak suggests a uniform size distribution and minimal aggregation, indicating effective nucleation and capping during the synthesis process. The convergence of the SPR peak positions in both methods implies that, despite potential differences in the synthetic protocols, the resultant nanoparticles share comparable physicochemical characteristics. However, UV/Vis spectroscopy alone cannot conclusively determine particle size or shape. The observed peak at 396 nm is consistent with the literature reports for silver nanoparticles synthesized via green or chemical reduction methods under controlled conditions. This wavelength lies well within the expected SPR window for colloidal AgNPs suspended in aqueous media and further supports the notion of a successful and reproducible synthesis process in both methods.
In Method 1, ultrasound energy plays a pivotal role in controlling the nucleation and growth of silver nanoparticles (AgNPs), primarily through the phenomenon of acoustic cavitation, the formation, growth, and implosive collapse of microbubbles in the liquid medium. This process generates localized hot spots with transient temperatures exceeding 5000 K and pressures above 1000 atm, along with intense shear forces and microjets. These extreme, localized conditions promote a rapid and homogeneous reduction of Ag+ ions, leading to a burst nucleation phase where a high number of nuclei form nearly simultaneously. This rapid nucleation depletes the local concentration of silver ions, thereby limiting the subsequent growth of individual nanoparticles and contributing to the formation of smaller and more uniformly sized particles. In contrast, Method 2, which lacks ultrasonic input, relies on passive thermal or chemical reduction, often resulting in less controlled nucleation, slower kinetics, and greater opportunity for particle growth and aggregation. Furthermore, the cavitational effects in Method 1 also help minimize nanoparticle aggregation by physically disrupting the forming clusters and by enhancing mixing at the molecular level. The high-frequency oscillations and microstreaming generated by ultrasound facilitate the uniform dispersion of nanoparticles throughout the reaction medium, thus stabilizing the colloid and preventing the coalescence of primary particles. These synergistic effects of ultrasound—accelerated nucleation, restricted growth, and anti-aggregation—explain the observed differences between the two methods and underline the effectiveness of ultrasound-assisted synthesis in producing well-defined AgNPs.
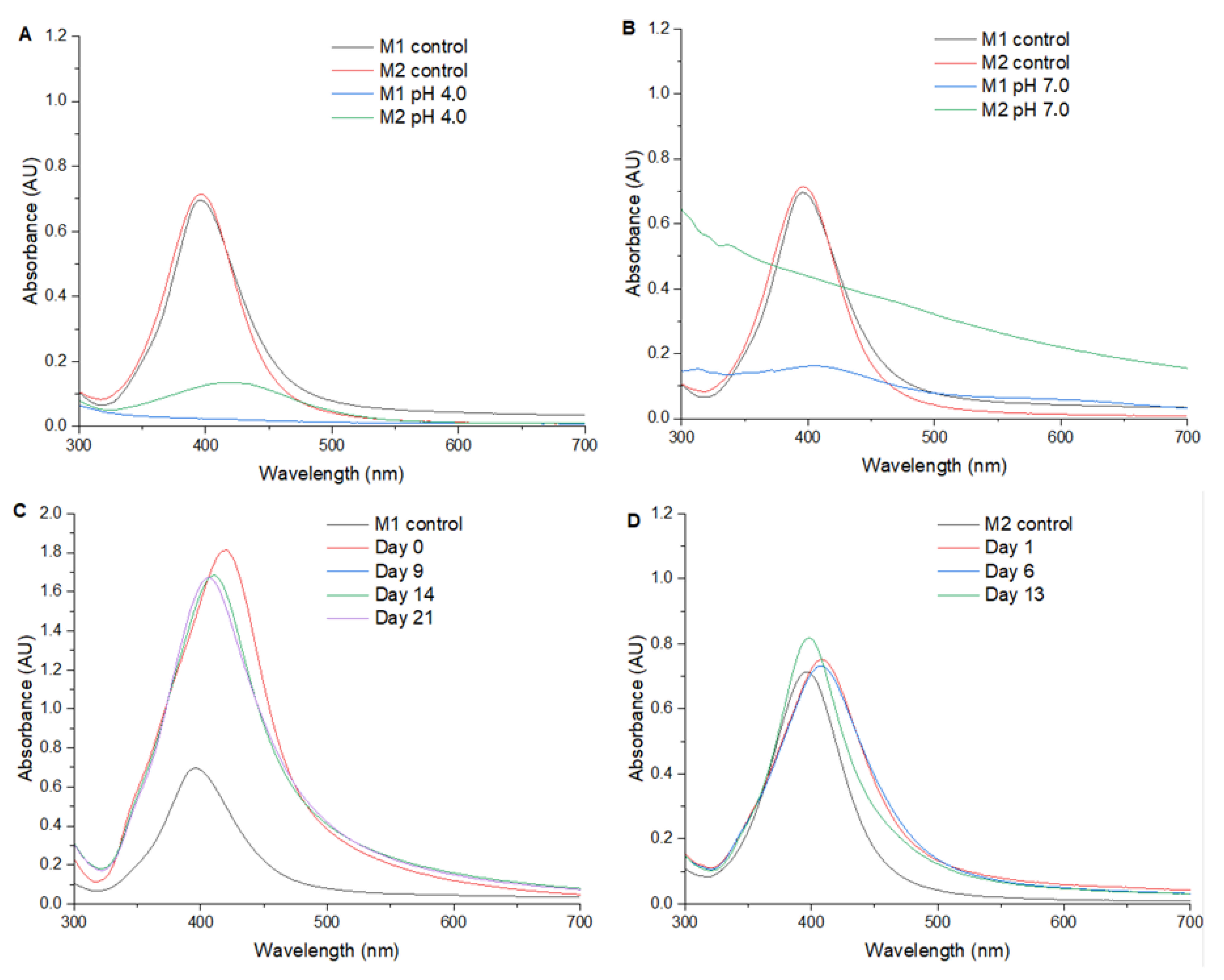
Figure 2 presents the UV/Vis spectra of AgNPs synthesized at various pH levels: 4.0; 7.0; 10.0; and a control sample in deionized water (neutral pH). This figure underscores the critical role of the reaction medium’s pH in determining the formation, size, and stability of silver nanoparticles.
At pH 4.0 and 7.0, no distinct SPR peak is observed within the 300–700 nm range, indicating that under these acidic and neutral conditions, the formation of AgNPs is either significantly hindered or results in particles that are poorly dispersed and/or irregular in shape. The absence of an SPR peak may also suggest the formation of aggregates or larger particles with broader size distributions that do not exhibit a well-defined plasmonic signature. One plausible explanation is that, under acidic conditions, the reducing power of the agents employed is suppressed due to proton competition, limiting the reduction of Ag+ to Ag0. Moreover, lower pH values may destabilize potential capping agents or prevent their effective interaction with the nanoparticle surface, leading to uncontrolled growth or precipitation.
In contrast, at pH 10.0, a clearly defined SPR peak appears at 406 nm, indicating successful nanoparticle formation. The 10 nm red shift in the SPR band compared with the neutral pH sample suggests an increase in nanoparticle size and/or a change in the dielectric environment surrounding the particles. Under alkaline conditions, the reduction of silver ions is typically more rapid, and the presence of excess hydroxide ions can facilitate the activation of functional groups in capping or stabilizing agents, enhancing nanoparticle stability and favoring more controlled growth kinetics.
This red shift may also reflect subtle changes in nanoparticle morphology (from perfectly spherical to slightly elongated or faceted shapes) as well as potential differences in the local refractive index caused by the nature of the stabilizing environment at higher pH.
The combined analysis of
Figure 1 and
Figure 2 highlights the dual importance of the synthesis methodology and the environmental pH in tailoring the optical and structural properties of AgNPs. The UV/Vis absorption spectra provide a rapid, non-destructive, and highly informative tool for monitoring nanoparticle formation, especially when correlated with complementary characterization techniques.
Notably, the presence, position, and shape of the SPR peak serve as proxies for assessing nanoparticle size, dispersion quality, and colloidal stability. The consistency of the 396 nm peak across two different synthesis methods underscores the robustness of the procedures used, while the significant spectral differences observed under variable pH conditions emphasize the necessity of careful pH control in nanoparticle synthesis protocols.
In practical terms, controlling pH during synthesis not only affects nanoparticle nucleation and growth kinetics but also plays a pivotal role in determining the final particle morphology and potential applicability in diverse fields. For example, AgNPs with narrower size distributions and strong SPR signals are particularly desirable for applications in sensing, diagnostics, and plasmonic devices, whereas larger or aggregated particles may be more suited for antimicrobial or catalytic applications.
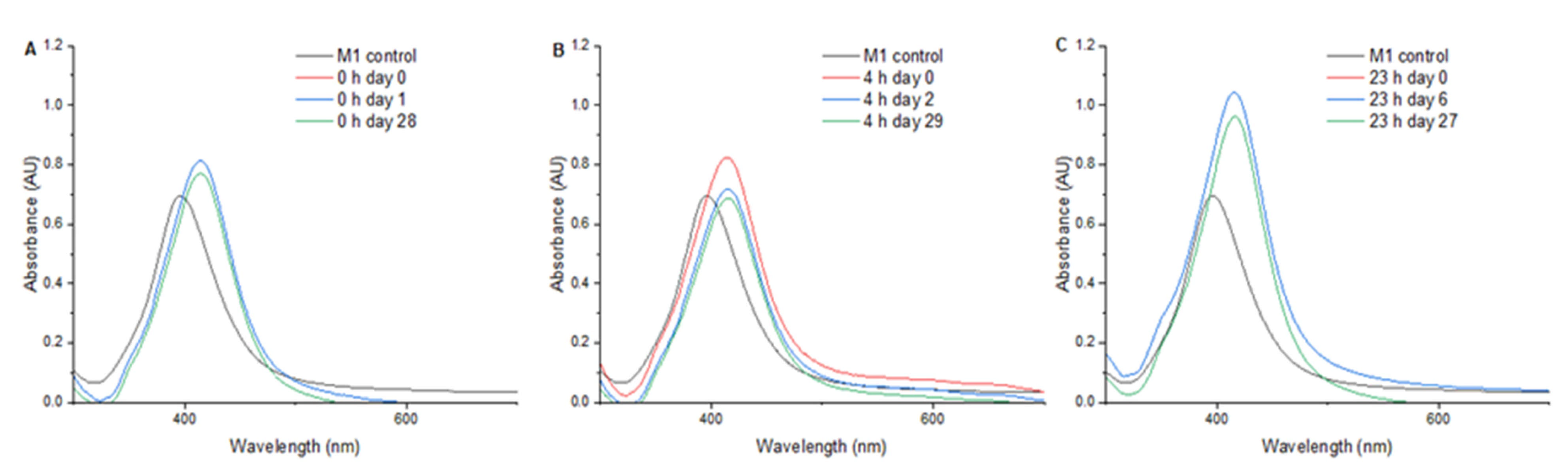
Figure 3 presents the UV/Vis spectra from 300 to 700 nm of AgNPs subjected to a pressure of 1.75 MPa for 0, 4, and 23 h. Upon treatment for 0 h (
Figure 3A), the AgNPs exhibited a maximum absorbance of 0.813 at 414 nm, showing an SPR shift of +18 nm. The measurement of SPR at one day and at 28 days after treatment showed no significant changes in the SPR. In the 4-h treatment (
Figure 3B), a maximum absorbance of 0.825 at 414 nm was observed, and a decrease in the SPR intensity was found two days after pressure treatment. In the 23-h treatment (
Figure 3C), an SPR shift of +19 nm and an increase in the plasmon peak intensity were found.
Table 1 summarizes the SPR maximum and SPR shift obtained for each AgNP treatment. Also, the “full width at half maximum” (FWHM) measured from the UV/Vis spectra is presented. Synthesis of AgNPs at pH 10 with Method 1 and the pressure treatment of AgNPs at 1.75 mPa for 23 h produced the more intense SPR (higher values). In contrast, Methods 1 and 2 produced sharper SPR peaks, which in turn suggest a more homogeneous size diameter distribution.
STEM images of AgNPs synthetized with the two synthesis methods are presented in
Figure 4.
Figure 4A presents the AgNPs synthetized with Method 1, showing spherical morphologies with a diameter distribution of 6 ± 2 nm.
Figure 4B presents the AgNPs synthetized with Method 2, which presents a diameter distribution of 21 ± 11 nm. AgNPs synthesized with Method 1 at pH 10.0 resulted in particles with spherical morphologies at a submicrometric scale with a diameter distribution of 300 ± 84 nm.
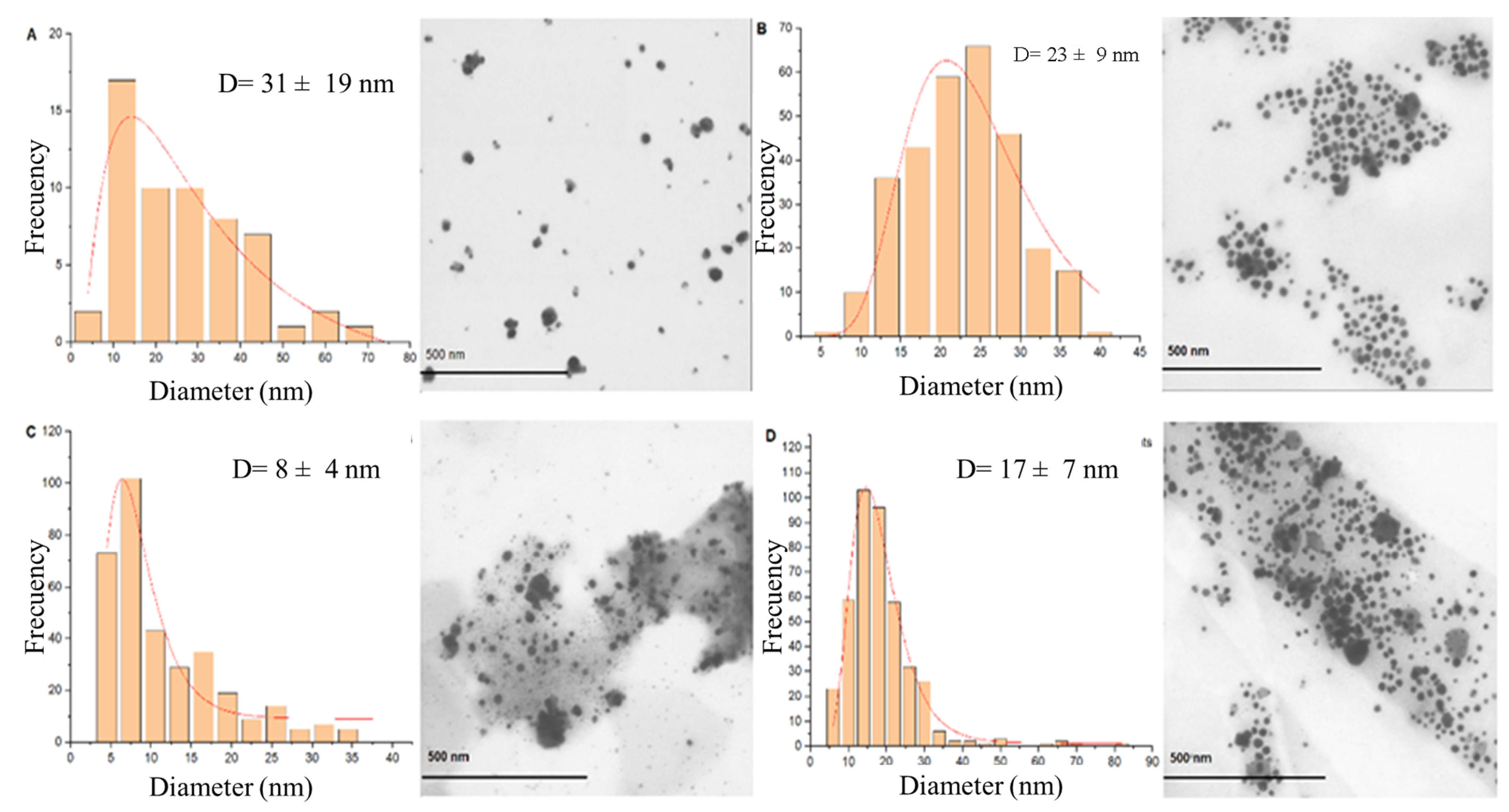
Figure 5 presents the synthesis with Method 2 at pH 10.0 with a size distribution of 31 ± 19 nm (
Figure 5A). The AgNPs obtained at the end of the synthesis and subsequently subjected to heat treatment at 200 °C until stabilization of the solvothermal reactor to 1.75 MPa (0 h) are presented in
Figure 5B. Very uniform spherical morphologies can be observed with a very symmetrical size distribution in a range of sizes of 23 ± 9 nm. At 4 h of pressure exposure (
Figure 5C), spherical AgNPs are observed with a size distribution of 8 ± 4 nm. At 23 h of pressure exposure (
Figure 5D), it is possible to observe spherical AgNPs with a much more uniform size distribution than the 0-h treatment and the 4-h treatment, with a predominance of AgNPs with a diameter in the range 17 ± 7 nm.
Due to the superior homogeneity and smaller particle size observed in silver nanoparticles (AgNPs) synthesized via Method 1, as indicated by their sharp and symmetric SPR peak, a representative sample from this method was selected for energy-dispersive X-ray spectroscopy (EDX) analysis (
Figure 6A). The EDX spectrum revealed the presence of several elements: oxygen (32%); sodium (31%); copper (17%); chlorine (11%); aluminum (3%); and silver (3%). While silver confirms the successful reduction of Ag
+ to metallic AgNPs, the presence of other elements can be attributed to multiple sources inherent to the synthesis and analysis process. The high oxygen content likely arises from surface-bound organic molecules used as reducing and capping agents as well as from the partial surface oxidation of AgNPs. Sodium may derive from residual reagents such as NaBH
4. The detection of copper is attributed to the copper grid commonly used in EDX sample preparation, which can contribute significantly to the elemental signal due to electron beam penetration. Similarly, the aluminum signal may originate from the sample stub or adhesive. Chlorine presence is likely due to residual chloride ions from reagents or the aqueous environment. These elements, therefore, do not necessarily represent contamination but reflect typical components of the synthesis medium, stabilizing matrix, and substrate materials, highlighting the complexity of nanoparticle characterization by EDX and the need to interpret such spectra in the context of both synthetic conditions and instrumentation artifacts. The elemental composition determined by EDX is further supported and enriched by Raman spectroscopy analysis (
Figure 6B), which provides complementary insights into the molecular and structural characteristics of the AgNPs synthesized via Method 1 under different conditions. Raman spectra were obtained for samples synthesized by the standard Method 1, Method 1 adjusted to pH 10, and Method 1 carried out under elevated pressure (1.75 MPa) for 4 h. In all cases, characteristic vibrational bands were observed corresponding to metallic Ag–Ag bonds, which confirm the presence of nanoscale silver clusters. Additionally, C–H stretching vibrations and Ag–O interactions were detected, suggesting the presence of organic capping agents or residual plant-derived compounds as well as the possible partial surface oxidation of the nanoparticles. Notably, in the sample subjected to high-pressure treatment, additional Raman bands associated with C–C bond vibrations were identified. These may reflect structural changes in the organic matrix, or the formation of new carbonaceous phases induced by prolonged exposure to pressure, with both possibly enhancing the interaction between organic stabilizers and the nanoparticle surface. The presence of these molecular features supports the complex chemical environment of the synthesized AgNPs and confirms the successful integration of metallic cores with stabilizing organic or oxide layers, which may play a crucial role in their stability and functional performance.
4. Discussion
4.1. Influence of the Synthesis Method on the Plasmonic Properties of AgNPs
The monitoring over time of AgNPs synthesized with Method 1 shows a greater stability of nanoparticles than AgNPs synthesized with Method 2, where an increase in the signal intensity occurs, suggesting that the AgNPs slightly agglomerate over time. In Method 2, it is observed that as time passes, a shift of the SPR occurs, attributed not only to the agglomeration of the nanoparticles but also to the formation of reactive forms of silver, such as the Ag
+ ion. Apparently, the dynamics of the AgNO
3 reduction reaction are different between the two methods. In Method 1, the addition of the mixture of NaBH
4 and Na-cit to the AgNO
3 solution favors the reduction of the AgNPs in a controlled manner, producing smaller, more uniform and stable AgNPs. In Method 2, the mixture of AgNO
3 with sodium citrate could cause an uncontrolled reduction when adding NaBH
4 due to the reducing effect of sodium citrate [
41,
42,
43,
44,
45], producing a greater heterogeneity in the size and shape of the NPs (
Figure 4B). This variability in morphology would contribute to a mild aggregation and an increase in the maximum of the SPR peak over time [
44].
4.2. Influence of pH on the Plasmonic Properties of Silver Nanoparticles
Regarding the pH effect on AgNP synthesis, at pH 4.0 (
Figure 2A), no SPR peak was detected. This finding suggests that the pH level of the reaction medium interferes with the formation and/or stability of the AgNPs nucleation, and it is not dependent on the method. The absence of an SPR peak at pH 4.0 suggests that the acidic environment interferes with the formation and stability of AgNPs, indicating that low pH conditions may slow down the reduction rate of silver nitrate, thereby altering the kinetics of AgNP synthesis. This observation is consistent with previous studies that have shown how acidic conditions can affect the reduction process and the resulting nanoparticle characteristics [
46,
47,
48,
49,
50].
At pH 7.0 (
Figure 2B), no SPR peak was found in the UV/Vis spectrum with both methods either. Considering that the synthesis of AgNPs in water (neutral pH) resulted in a defined SPR, it could be assumed that the ionic strength generated by the 20 mM phosphate buffer solution at pH 7.0 could be interfering with the reduction of AgNO
3 or nucleation of AgNPs, highlighting the critical role of buffer composition and concentration in the synthesis process.
Regarding the synthesis at pH 10.0, it is observed that Method 1 (
Figure 2C) produces a more intense peak of the SPR compared with Method 2 (
Figure 2D) and with Method 1 in water (
Figure 1A), suggesting a higher efficiency in the synthesis of AgNPs in alkaline medium. In addition, the SPR presented a shift of +22 nm in contrast to Method 1 in water. Time–course monitoring of AgNPs shows great stability for both methods, with Method 1 maintaining a higher SPR intensity than Method 2.
At pH 10.0, the presence of a more intense SPR peak with Method 1 compared with Method 2 and Method 1 in water, indicates that an alkaline environment enhances the synthesis efficiency of AgNPs. The observed shift in SPR (>22 nm) suggests changes in the size and morphology of the nanoparticles, attributable to the higher reduction rate of silver nitrate in basic conditions. These results demonstrate that the efficiency of AgNP synthesis is method dependent, with Method 1 showing higher SPR intensity and stability compared with Method 2. have suggested that pH conditions exert a significant impact on the size and stability of AgNPs, with higher pH values favoring the stability and growth of nanoparticles [
46,
47,
48,
49,
50].
The absence of observable SPR peaks at pH 4.0 and 7.0 (
Figure 2B) is indeed noteworthy and was initially attributed to ionic strength effects and reduced reduction kinetics; however, a more detailed mechanistic explanation is warranted. At pH 4.0, where a citrate buffer is typically employed, citrate molecules are predominantly in their protonated form, reducing their ability to act as effective reducing and capping agents. More importantly, citrate is known to function as a chelating agent, especially under acidic conditions, forming stable complexes with metal ions such as Ag
+. This chelation may significantly sequester free silver ions in solution, thereby inhibiting their reduction and delaying or entirely suppressing the nucleation of silver nanoparticles. This mechanism has been proposed in previous studies where citrate, at low pH, acts more as a complexing agent than as a reducing agent, preventing the supersaturation necessary for nucleation. At neutral pH (7.0), citrate exists in a partially deprotonated state and may still exhibit moderate chelating behavior, which could similarly limit the availability of free Ag
+ ions. Additionally, the buffering capacity and ionic environment at this pH may reduce the activity of reducing agents, thereby slowing the reduction kinetics below the threshold needed for effective nanoparticle formation. These pH-dependent interactions, including Ag
+–citrate complexation and pH-sensitive protonation states of organic ligands, appear to be central to the observed suppression of SPR signals and highlight the crucial role of buffer chemistry in nanoparticle synthesis.
In this study, the influence of pH on the synthesis of silver nanoparticles (AgNPs) was thoroughly examined, and it was found to play a critical role in determining both the size and morphology of the nanoparticles. The pH level affects the reduction rate of the precursor as well as the stability of the nanoparticles and their final size distribution. At acidic pH values (below 7.0), the reaction medium’s conditions hinder the reduction process of silver ions largely due to the lower reduction rates and the destabilizing effect of salts in the medium. These conditions impede the effective formation of AgNPs, often resulting in irregular shapes, aggregation, or even a complete lack of nanoparticle formation. The presence of salts at acidic pH levels can also lead to the formation of silver–salt complexes, which can reduce the efficiency of the reducing agents, making it more difficult to achieve a controlled synthesis of AgNPs.
In contrast, at alkaline pH values (above 7.0), particularly at pH 10.0, the reduction rate of silver ions is significantly accelerated, leading to a more efficient synthesis of AgNPs. In these conditions, the nanoparticles formed are generally larger, with diameters exceeding the typical nanometric scale. This phenomenon is accompanied by noticeable shifts in the surface plasmon resonance (SPR) peaks, which are indicative of changes in the optical properties of the AgNPs. The SPR shift suggests that larger particles with altered optical behavior are formed under alkaline conditions, which could be beneficial for applications that rely on specific optical characteristics of the nanoparticles, such as plasmonic sensing and optical imaging. Furthermore, the increase in nanoparticle size at higher pH values could affect their stability and reactivity, influencing their potential use in fields like biosensing and nanomedicine, where precise control over nanoparticle properties is essential.
4.3. Influence of Pressure on the Plasmonic Properties of Silver Nanoparticles
The selection of 1.75 MPa and 200 °C for the pressure-assisted synthesis of silver nanoparticles (AgNPs) was based on a combination of preliminary optimization trials and precedents in the literature indicating that moderate hydrothermal conditions can significantly influence nanoparticle growth, crystallinity, and surface chemistry. While there is no universal standard for pressure–temperature parameters in nanoparticle synthesis, values in the range of 1–3 MPa and 150–250 °C have been widely reported in hydrothermal or solvothermal methods, particularly for controlling particle morphology and enhancing crystallinity. It has been demonstrated that silver nanoparticles synthesized at ~2 MPa and 180 °C exhibited improved size uniformity and reduced aggregation compared with those synthesized at ambient conditions. Similarly, it has been reported that hydrothermal treatments around 200 °C favored the formation of highly crystalline AgNPs with superior optical properties. In our study, 1.75 MPa and 200 °C were chosen as intermediate values to balance particle stabilization with energy efficiency and to avoid excessive sintering or particle coalescence, which are common at higher pressures and temperatures. Although further systematic optimization could be performed, our preliminary findings suggest that these conditions are effective in modifying the nanoparticle environment—evidenced by the appearance of additional Raman bands (e.g., C–C bonds)—and in enhancing the structural integrity of the AgNPs. Future studies will aim to fine-tune these parameters more precisely and to explore their effects across a broader range of characterization techniques.
In the 0-h treatment, a shift in the SPR peak of +18 nm was found, and the AgNPs presented great stability at one day and at 28 days after treatment, suggesting that the pressure applied after synthesis promoted the formation of stable and well-defined structures. The 4-h treatment also presented a +18 nm shift in SPR but led to a higher absorbance and a decrease in SPR intensity over time, indicating potential instability or morphological changes.
For the 23-h treatment, the SPR peak presented a shift of +19 nm and an increase in the plasmon peak, as shown in
Figure 3C, which indicates that an increased consolidation or growth of the nanoparticles occurred. The shift in SPR may be caused by changes in the crystal structure or by the agglomeration of nanoparticles. Previous findings have shown that the application of high pressures on nanoparticles induces significant changes in their morphology and plasmon resonance, attributed to recrystallization or changes in the crystal structure due to rearrangement of atoms under high pressure [
51,
52,
53] The observed shift in the SPR maximum following pressure treatment indicates significant alterations in the optical properties of the AgNPs. These observations are consistent with prior research, which shows that high pressure can induce significant changes in nanoparticle morphology and optical properties through recrystallization or atomic rearrangement [
54].
The SPR peaks and their corresponding FWHM values are summarized in
Table 1 for pH, method, and pressure treatments and show how Methods 1 and 2 present the lower FWHM values, indicating a sharper SPR than at pressure treatment or at an alkaline pH level. Despite this, the SPR peak is higher when AgNPs are subjected to pressure and a pH of 10.0. The FWHM is a measure of sharpness of the SPR, which in turn is related to NP diameter and the precision of shift determination in the SPR peak, leading to a higher sensitivity in detecting molecular binding events on the NP surface [
49]. According to this, pressure treatments and pH levels are interesting ways by which to modify or synthesize AgNPs due to their higher SPR peaks and acceptable sharpness.
In addition to pH, the application of pressure during the synthesis process was found to have a significant impact on the size, uniformity, and morphology of the AgNPs. Pressure treatment promotes the nucleation and growth of nanoparticles by increasing the local temperature and enhancing the reduction rate of the precursor. High pressure leads to an increase in the size of the AgNPs, which is beneficial when the synthesis of larger nanoparticles is desired. More importantly, pressure also improves the uniformity of the nanoparticles, resulting in a more monodispersed particle population. This is a key advantage for applications that require nanoparticles with consistent size and shape, such as biosensors and catalysis, where uniform particle characteristics are crucial for optimal performance.
The pressure-induced changes in size and morphology are likely due to the enhanced controlled nucleation and growth of AgNPs under pressure, which prevents the aggregation of particles and promotes the formation of more ordered crystalline structures. These findings are particularly important for applications that require nanoparticles with tight size distributions, as uniformity can greatly enhance the sensitivity and efficiency of catalytic reactions. In biosensing, for example, uniform nanoparticles lead to more reliable and reproducible results, as the interactions between the nanoparticles and target molecules become more consistent. Similarly, in catalysis, where nanoparticle size is closely related to the surface area, having well-controlled particles ensures maximum catalytic activity and efficiency.
Moreover, prolonged pressure exposure not only stabilizes the nanoparticles but also facilitates the formation of more ordered crystalline structures, contributing to a tighter size distribution and improved morphological uniformity. The resulting nanoparticles exhibit enhanced optical and catalytic properties, which are crucial for achieving high-performance systems in fields like surface-enhanced Raman spectroscopy (SERS), electrochemical sensing, and environmental remediation.
4.4. Characterization of the Size and Functional Groups of Silver Nanoparticles
STEM images allowed us to confirm that the uncontrolled reduction of AgNO
3 caused by the mixture containing Na-cit produces larger particles with Method 2. In the same way, the shift caused by Method 1 at pH 10 is caused by the larger size of the NPs. In this respect, the development of three different synthesis methods allowed us to control the size and distribution of AgNPs, producing very small AgNPs with Method 1, followed by Method 2, and the largest NPs with Method 1 at pH 10.0. The advantage of producing large AgNPs with the higher SPR peak and an acceptable FWHM is related to the advantage of producing AgNPs with larger diameters, favoring the number of capping molecules, with the disadvantage of restricting the access of counterions near the thiol-modified AuNPs in contrast to the smaller AuNPs [
54].
Although AgNP size increases according to the method applied, the use of ultrasound energy promotes the production of small particles in the case of Method 1 and allows the control of size in Method 2 due to the Na-cit reducing effect. These insights highlight the importance of utilizing ultrasound as a rapid and cost-effective method for synthesizing silver nanoparticles, as it is employed for the biological synthesis of AgNPs. Ultrasound offers an efficient means to control particle size and distribution while minimizing synthesis time and cost. This technique provides a valuable alternative for producing AgNPs with precise characteristics, ensuring high quality and performance for a range of applications. The integration of ultrasound in the synthesis process not only enhances the feasibility of nanoparticle production but also supports the development of scalable and economically viable nanotechnology solutions.
In the same way, the heat treatment for 0 h produced a range of sizes of 23 ± 9 nm, suggesting that the size of the nanoparticles is affected due to the exerted pressure of 1.75 MPa and the exposure time, in comparison with water synthesis, suggesting that the size of the nanoparticles is affected due to the exerted pressure of 1.75 MPa and the exposure time. Note that before subjecting the AgNPs to the hydrothermal process, the average size is small (6 nm), but after pressure exposure, the size of the AgNPs increases (17 nm). According to these results, prolonged pressure at 1.75 MPa allows the production of very uniform AgNPs of larger size compared with Method 1 and Method 2 synthesis at atmospheric pressure. Crystal formation was also observed for 4 and 23 h of pressure exposure. Therefore, the increase in time exposure to pressure allows the formation of more homogeneous and larger AgNPs.
The influence of pressure on the larger size of AgNPs and on uniform morphology can be influenced by different factors. Apparently, the nucleation and growth of AgNPs is favored and influenced by constant pressure. In consequence, the morphology of AgNPs is more uniform and the size distribution is narrower. A sustained pressure for a prolonged period has a synergistic effect on the growth of the nanoparticles, as observed in the increase in size from 0 to 23 h. Yang et al. [
48] found that hydrothermal synthesis of AgNPs at 100 °C for 6 h produced spherical particles of 15 nm on average, but synthesis for 12 h produced irregular particles of 10–40 nm. Increasing the temperature to 180 °C for 6 h produced truncated triangular nanosheets; meanwhile, 12-h treatment disk-like nanoparticles were obtained [
55,
56,
57,
58,
59,
60,
61,
62,
63] reported the presence of rods and plates, but the use of chitosan as a dispersant apparently aggregated the AgNPs when subjected to hydrothermal treatment at 100 °C and 150 °C.
Integrating these findings with the observed variations in size, pressure treatment significantly impacts both the size and uniformity of AgNPs as well as their chemical composition and structural characteristics. The presence of graphite, coupled with changes in elemental composition, illustrates the complex effects of pressure on the synthesis of nanoparticles. This complex interplay between physical conditions and chemical composition affects critical aspects such as the SPR shift, which reflects alterations in the nanoparticle’s electronic environment. For instance, the observed SPR shift of +20 nm with pressure treatment indicates notable changes in the electronic structure and morphology of the AgNPs, which are directly related to the formation of more uniform and larger particles. The impact of pressure on AgNPs not only underlines the requirement of optimal synthesis parameters to achieve specific nanoparticle properties but also emphasizes the critical role of such optimization for diverse applications. In fields such as catalysis, the size, the shape, and the chemical environment of nanoparticles are pivotal for their efficiency and effectiveness. Understanding and controlling these parameters enables researchers to tailor AgNPs for enhanced performance in various applications from environmental sensing to advanced materials.
Finally, the elemental composition of AgNPs indicates the presence of oxygen and sodium, which corroborates the presence of sodium citrate and sodium borohydride used in AgNP synthesis and likely to some degree the oxidation of the nanoparticles. The detected copper and aluminum are related to the matrix on which the sample was analyzed; meanwhile, chlorine is derived from ammonium chloride in the buffer solution. Silver constitutes only 3% of the total composition, reflecting the relative abundance of the silver nanoparticles within the sample. The Raman spectroscopy confirms the presence of silver nanoparticles, as based on the detected Ag–Ag bonds, and confirms the presence of sodium citrate (organic stabilizers), as based on the detected C–H bonds from methyl groups. Additionally, the detection of Ag–O bonds suggests that the nanoparticles have undergone mild oxidation. The detected C–C bonds indicate the formation of graphite in its two forms—D-graphite (amorphous) and G-graphite (ordered). This formation of graphite is a result of the high-pressure environment affecting the carbon components of citrate, leading to a more complex and ordered carbon structure [
55,
56,
57,
58,
59,
60,
61,
62,
63].






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}