
An Efficient Electroporation Protocol Supporting In Vitro Studies of Oligodendrocyte Biology


Abstract
1. Introduction
2. Experimental Design
2.1. Materials
- DMEM/F12 (ThermoFisher Scientific, Waltham, MA, USA, Cat. no.: 10565018)
- Neurobasal medium (ThermoFisher Scientific, MA, USA, Cat. no.: 12348017)
- Opti-MEM I Reduced Serum Medium (ThermoFisher Scientific, MA, USA, Cat. no.: 31985062)
- Hanks’ Balanced Salt Solution without Calcium and Magnesium (HBSS(−)) (ThermoFisher Scientific, MA, USA, Cat. no.: 14175053)
- HBSS with Calcium and Magnesium (HBSS(+)) (ThermoFisher Scientific, MA, USA, Cat. no.: 14025092)
- Poly-L-Ornithine (PLO) (Sigma, St. Louis, MO, USA, Cat. no.: A-004-M)
- Matrigel growth factor reduced (MG-GFR) (Corning, NY, USA, Cat. no.: 354230)
- Trace Elements B (1000×) (Corning, NY, USA, Cat. no.: 25-022-CI)
- B27 supplement (50×) (ThermoFisher Scientific, MA, USA, Cat. no.: 17504044)
- Platelet-derived growth factor (PDGF-AA) (Funakoshi, Tokyo, Japan, Cat. no.: 100-13A)
- Ciliary neurotrophic factor (CNTF) (Funakoshi, Tokyo, Japan, Cat. no.: 450-13)
- Neurotrophin-3 (NT3) (Funakoshi, Tokyo, Japan, Cat. no.: 450-03)
- Triiodothyronine (T3) (Sigma, MO, USA, Cat. no.: T6397)
- Nerve growth factor (NGF) (Merck, Darmstadt, Germany, Cat. no.: 01-125)
- 5-Fluoro-5′-deoxyuridine (FdU) (Sigma, MO, USA, Cat. no.: F8791)
- N-Acetyl-L-cysteine (NAC) (Sigma, MO, USA, Cat. no.: A9165)
- Isoflurane (FUJIFILM, Osaka, Japan, Cat. no.: 099-06571)
- 4% Paraformaldehyde Phosphate Buffer Solution (4% PFA) (FUJIFILM, Osaka, Japan, Cat. no.: 161-20141)
- Neural Tissue Dissociation Kit (P) (NTDK (P)) (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. no.: 130-092-628)
- CD140a (PDGFRα) MicroBead kit (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. no.: 130-101-502)
- Dead Cell Makeup Deep Red (DOJINDO, Kumamoto, Japan, Cat. no.: C556)
- AA3-PLP/DM20 (PLP) antibody [24]
- βIII-Tubulin antibody (Sigma, MO, USA, Cat. no.: T8578)
- GFP antibody (Abcam, Cambridge, UK, Cat. no.: ab13970)
- Olig2 antibody (Merck, Darmstadt, Germany, Cat. no.: MABN50)
- PiggyBac Transposon Vector (Funakoshi, Tokyo, Japan, Cat. no.: PB530A-2)
- Super PiggyBac Transposase Expression Vector (Funakoshi, Tokyo, Japan, Cat. no.: PB210PA-1)
2.2. Equipment
- MS columns (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. no.: 130-042-201)
- MiniMACS Separator (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. no.: 130-042-102)
- MACS MultiStand (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat. no.: 130-042-303)
- Super Electroporator NEPA21 Type II (NEPA21) (NEPAGENE, Chiba, Japan)
- Nepa Electroporation Cuvettes 2 mm Gap (NEPAGENE, Chiba, Japan, Cat. no.: EC-002)
- Cuvette chamber (NEPAGENE, Chiba, Japan, Cat. no.: CU500)
- 13 mm cover glass (MATSUNAMI, Osaka, Japan, Cat. no.: C013001)
- 4 well multi-dish (ThermoFisher Scientific, MA, USA, Cat. no.: 176740)
- Pasteur pipette (AS ONE, Osaka, Japan, Cat. no.: 2-2045-01)
- Mini Cell Strainer II 40 μm (Funakoshi, Tokyo, Japan, Cat. no.: HT-AMS-04002)
- All-in-one fluorescence microscopes (KEYENCE, Osaka, Japan, Cat. no.: BZ-X810)
3. Procedure
3.1. Preparation of Cover Glasses and NTDK (P) Reagents
- Place 13 mm cover glasses in each well of 4-wells chamber.
- Coat with 350 μL of PLO at 37 °C for overnight.
- Remove the PLO, rinse with distilled water twice, and dry the cover glasses.
- Coat the cover glasses with Matrigel GFR (MG-GFR) and incubate in a CO2 incubator for ≥30 min.
- Prepare Enzyme Mix 1 and Enzyme Mix 2 of NTDK (P) as described in the manual (Table 1).
- Warm 500 μL Enzyme Mix 1 in a 1.5 mL tube at 37 °C, and keep Enzyme Mix 2 on ice until use.
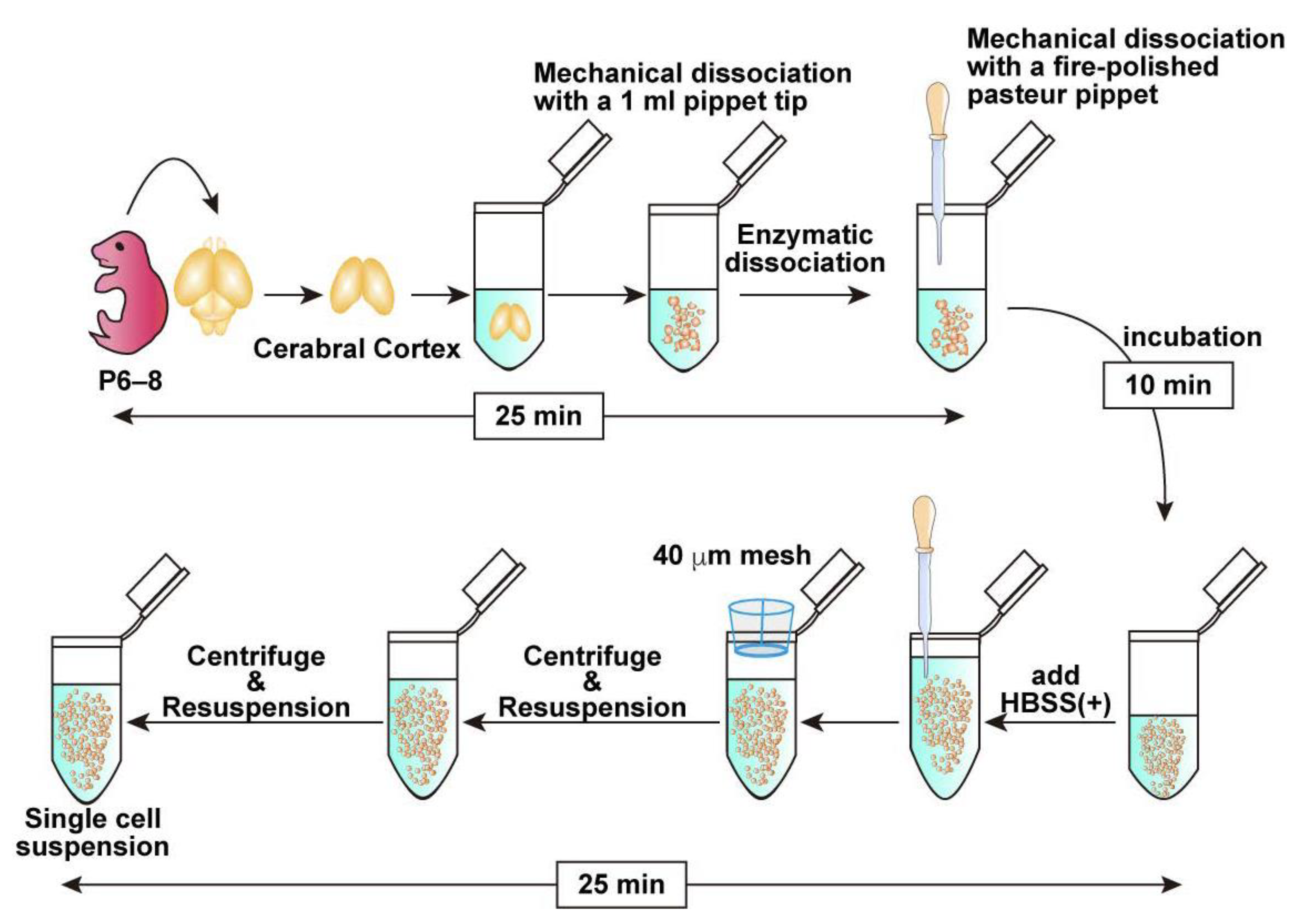
3.2. Brain Collection and Cell Dissociation (See Figure 1; Estimated Time: 60 min)
- Anesthetize a postnatal day 6–8 (P6–8) mouse with isoflurane and sterilize its body using 70% ethanol.Note: P6–8 mice are commonly used in this protocol because their tissue is easier to handle and typically yields sufficient numbers of OPCs. While younger mice can also be used, efficiency tends to decrease in older animals beyond this age.
- Decapitate and collect the brain in cold HBSS (−).
- Carefully dissect and collect the cortex. If possible, remove the meninges.Note: Meningeal removal is optional. Retained meninges do not affect the OPC purification efficiency. While removal of the meninges is preferable when feasible, prolonged efforts to remove them may reduce the overall cell yield. Therefore, if meningeal removal is perceived to be time-consuming, it is advisable to proceed promptly with tissue dissociation to preserve cell viability.
- Transfer the collected brain tissue into 500 μL of pre-warmed Enzyme Mix 1 prepared in a 1.5 mL tube. Pipette up and down with a 1 mL pipette tip 7–10 times to mince the tissue into small pieces approximately 1 mm in size.
- Incubate in the tube for 10 min at 37 °C.Note: Although rotation is recommended by the manufacturer’s protocol, it is not required under the present conditions.
- Add 7.5 μL Enzyme Mix 2, tap the tube to briefly mix the solution, dissociate the tissue mechanically with a fire-polished Pasteur pipette and incubate at 37 °C for an additional 10 min.Note: Enzyme Mix 2 must be mixed before mechanical dissociation as the tissue solution tends to be very sticky because of the release of genomic DNA from dead cells. Complete dissociation is not required in this step. Remaining Enzyme Mix 2 can be stored at 4 °C and retains enzymatic activity for at least 1 month.
- Add 750 μL HBSS (+). Then, further dissociate the tissue into smaller pieces by gently pipetting up and down into a single-cell suspension using a new fire-polished Pasteur pipette.
- Pass half of the cell suspension through a 40 µm cell strainer into a new 1.5 mL tube to obtain a single-cell suspension. Subsequently, apply 700 μL of HBSS (+) to the strainer to collect any remaining cells on the mesh. Repeat the same procedure with the remaining half of the cell suspension, collecting in a separate 1.5 mL tube.Note: If approximately 1.0 × 105 cells are sufficient for downstream applications, the second filtration step may be omitted.
- Centrifuge the cell suspension at 400× g for 10 min at 4 °C and remove the supernatant carefully.
- Resuspend the cell pellet in 1 mL of HBSS (+), then repeat the centrifugation under the same condition, and remove the supernatant carefully.
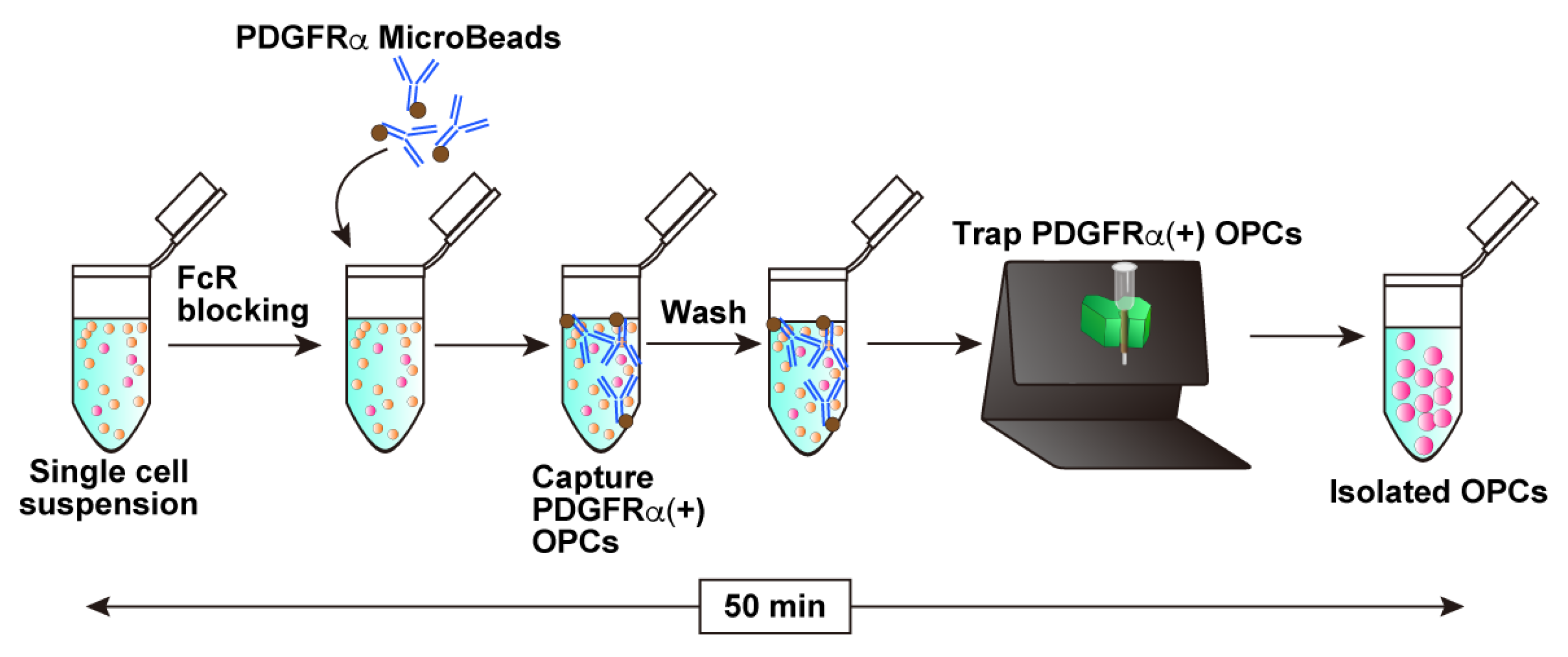
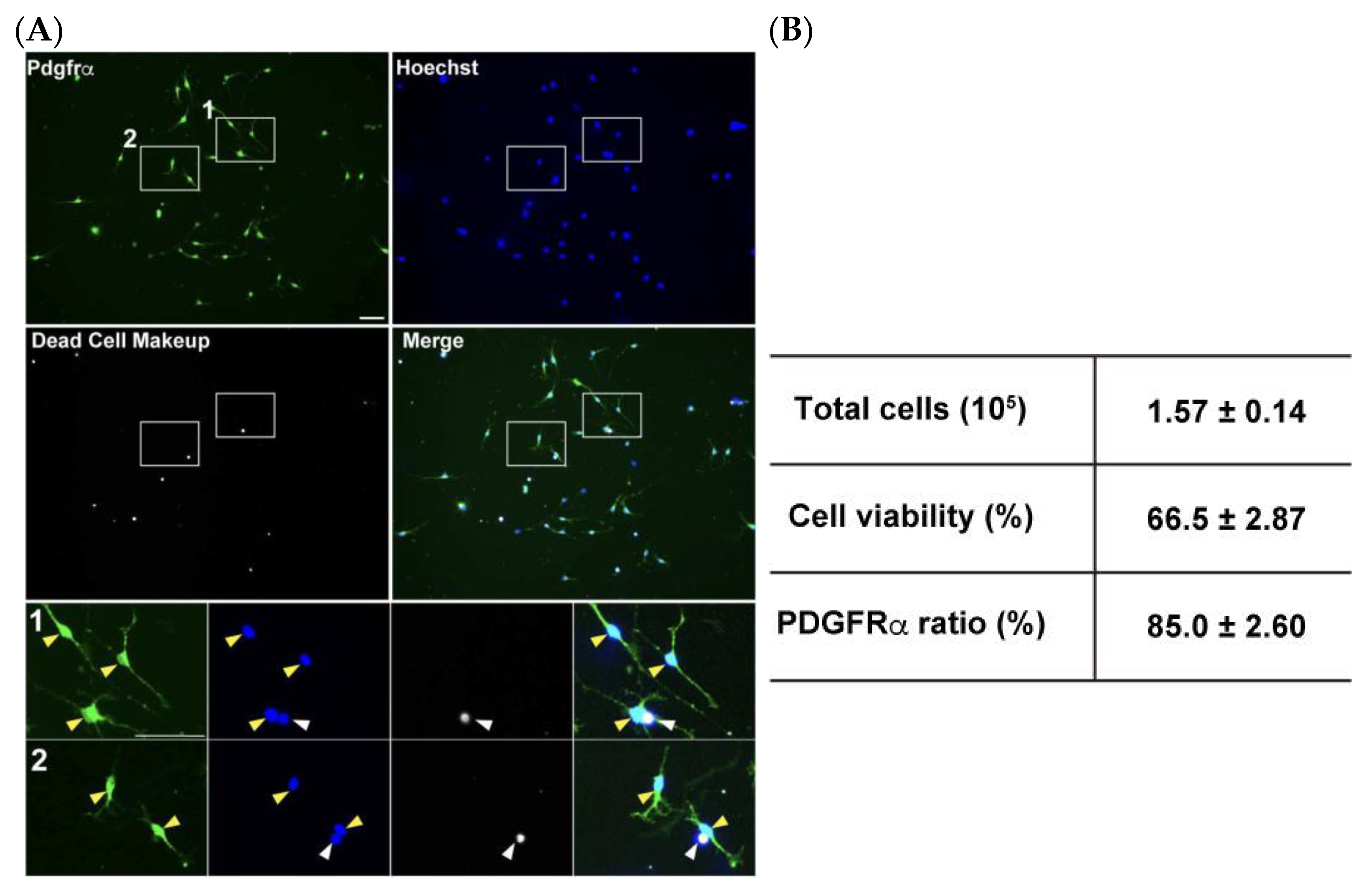
3.3. Magnetic Sorting with CD140a (PDGFRα) Microbead Kit (See Figure 2; Estimated Time: 50 min)
- Resuspend the cell pellet in 80 μL of PBS containing 0.5% BSA (0.5% BSA/PBS) and add 7 μL of FcR Blocking Reagent supplied in the CD140a (PDGFRα) Microbead kit.
- Incubate for 10 min at 4 °C.
- Add 7 μL of CD140a (PDGFRα) MicroBeads and mix gently by pipetting.
- Incubate for 10 min at 4 °C.
- Wash the cell suspension by adding 1.2 mL of 0.5% BSA/PBS and centrifuge at 500× g for 10 min.
- Remove the supernatant carefully and resuspend the cell pellet in 500 μL of 0.5% BSA/PBS.
- Proceed to the following magnetic isolation procedure.
- Place an MS column onto a MACS separator settled on a MACS Multistand.Note: MS columns are suitable for processing small volumes of cell suspensions. Prepare one MS column for one 1.5 mL cell suspension.
- Apply 500 μL of 0.5% BSA/PBS to the column and allow the buffer to pass through completely.
- Apply the whole cell suspensions to the column.
- Wash the column with 500 μL of 0.5% BSA/PBS 3 times.
- Remove the column from the MACS separator, apply 1 mL of Opti-MEM to the column, and collect the magnetically retained PDGFRα-positive cells by firmly plunging it into a 1.5 mL tube.Note: If the isolated cells are cultured directly without electroporation, use the culture medium for cell elution.
- Count the collected cells. Typically, 1.0–2.0 × 105 cells in total would be collected.Note: This cell number is from one MS column, meaning that 2.0–4.0 × 105 cells would be collected from one mouse brain since cell suspensions were split into two 1.5 mL tubes. However, applying cell suspensions from the two split tubes to one MS column does not increase the total cell number for recovery.
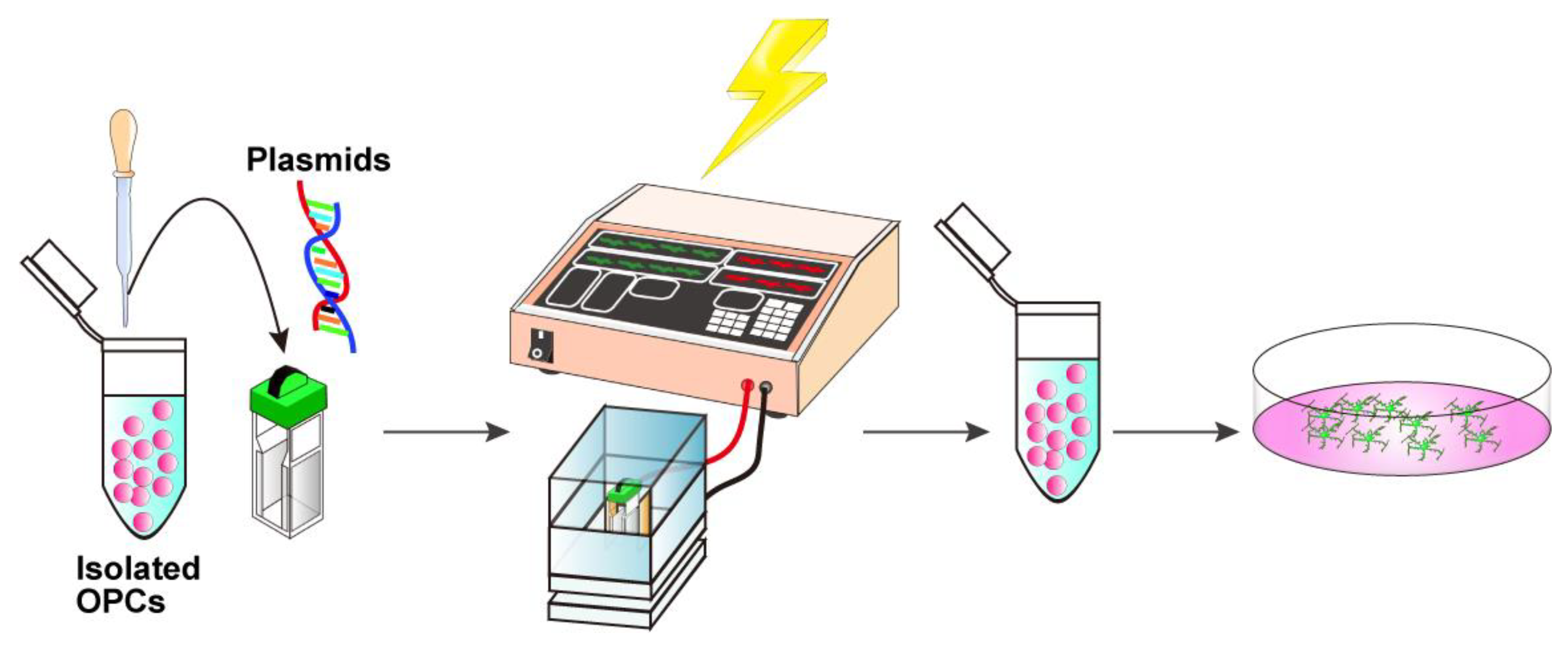
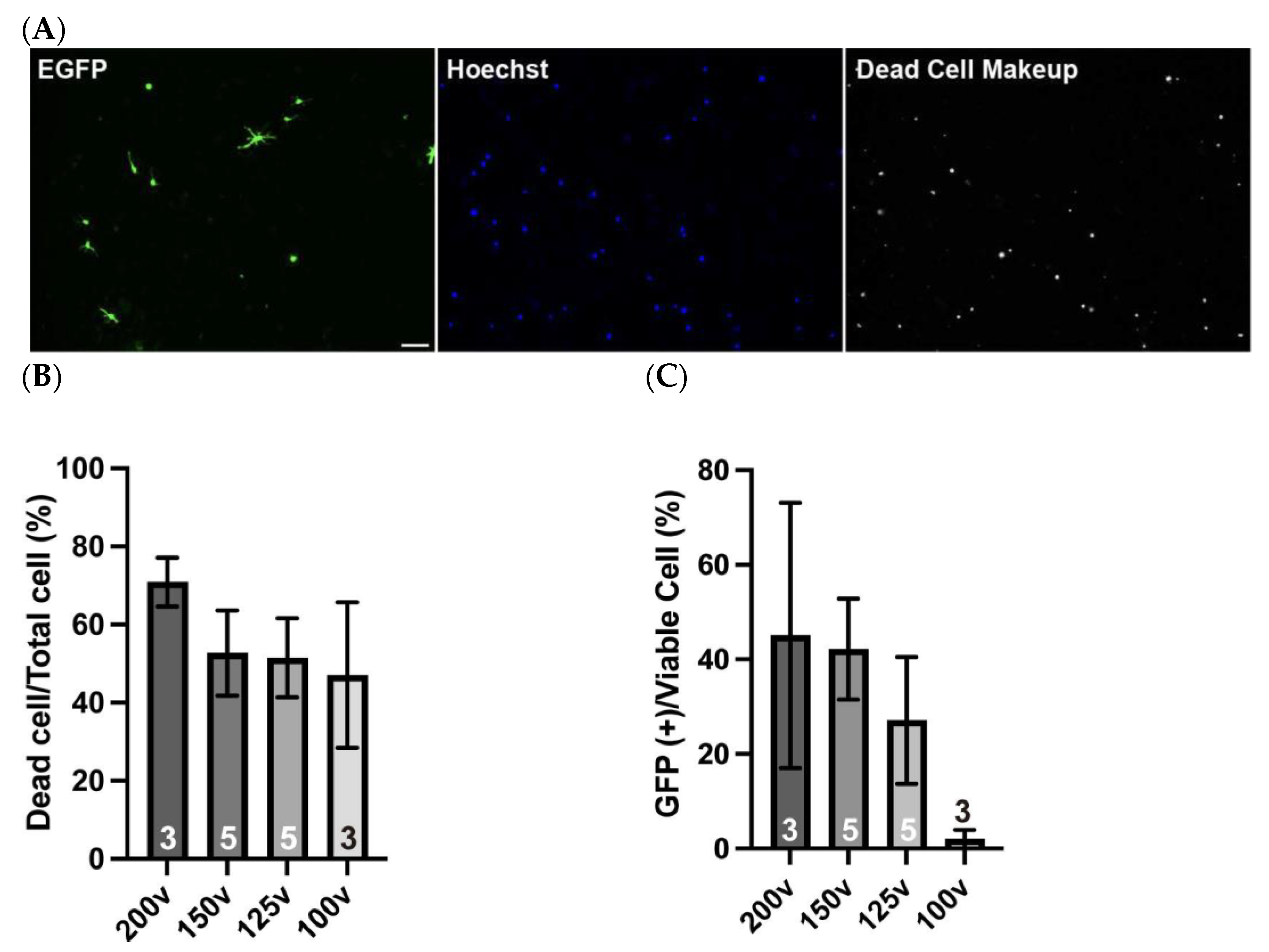
3.4. In Vitro Electroporation with a NEPA21 Electroporator (See Figure 3; Estimated Time: 10 min)
- Centrifuge the isolated cells at 500× g for 10 min at 4 °C. Discard the supernatant and gently resuspend the cell pellet in Opti-MEM at a density of 5 × 104 cells per 90 μL.Critical: At this step, the Opti-MEM should be kept at room temperature because the chilled medium may cause condensation on the cuvette surface, which can reduce the electroporation efficiency.
- Add 10 μg of DNA solution to 90 μL cell suspension, adjust the total volume to 100 μL with Opti-MEM, mix gently with a fire-polished Pasteur pipette, and transfer the entire cell–DNA mixture into an electroporation cuvette.Critical: Ensure that no bubbles are present in the cuvette. If bubbles are present, gently tap the cuvette until they disappear.
- Firmly insert the cuvette into the cuvette chamber.
- Perform electroporation using the NEPA21 system under the following conditions:Poring pulse, 150 V; pulse length, 1 ms; interval, 50 ms; number of pulses, 2; decay rate, 10%; pulse direction switch, none.Transfer pulse, 30 V; pulse length, 50 ms; interval, 50 ms; number of pulses, 5; decay rate, 50%; pulse direction switch, yes.
- Immediately transfer the transfected cells into 500 μL of OPC proliferation medium (Table 2).Optional: Debris can be removed by passing the electroporated cell solution through a 40 μm mesh. However, a significant number of cells may be lost due to their retention on the mesh.
- Plate the cells onto PLO/MG-GFR-coated 13 mm cover glasses placed in a 4-well multi-dish and culture in the OPC proliferation medium.
- For OPC differentiation, replace the OPC proliferation medium with OPC differentiation medium (Table 2) on day 2. Replace half of the medium every other day until analysis.
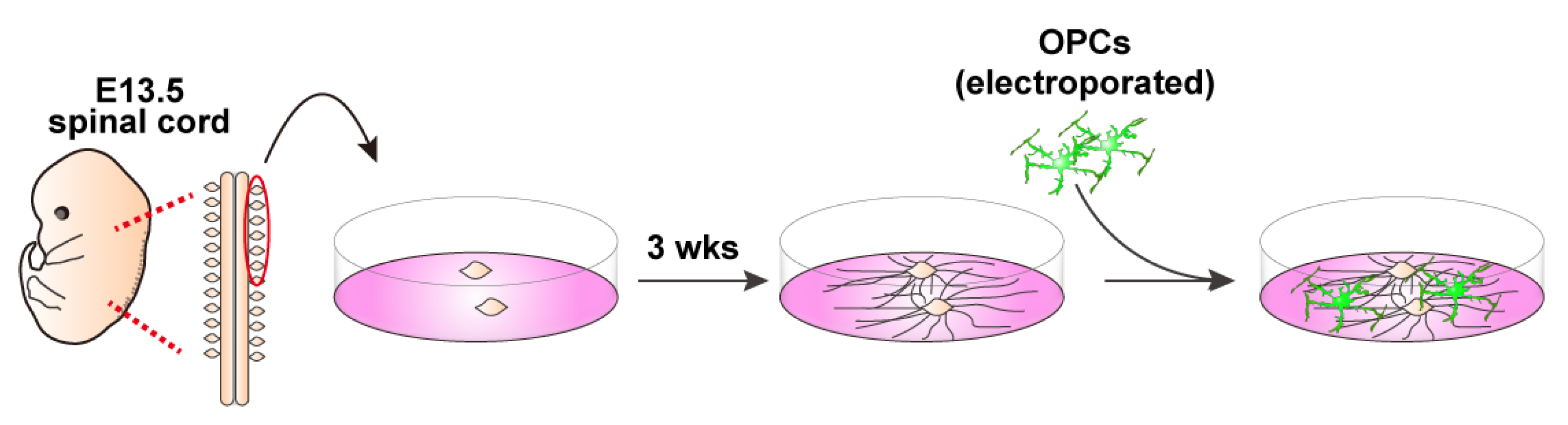
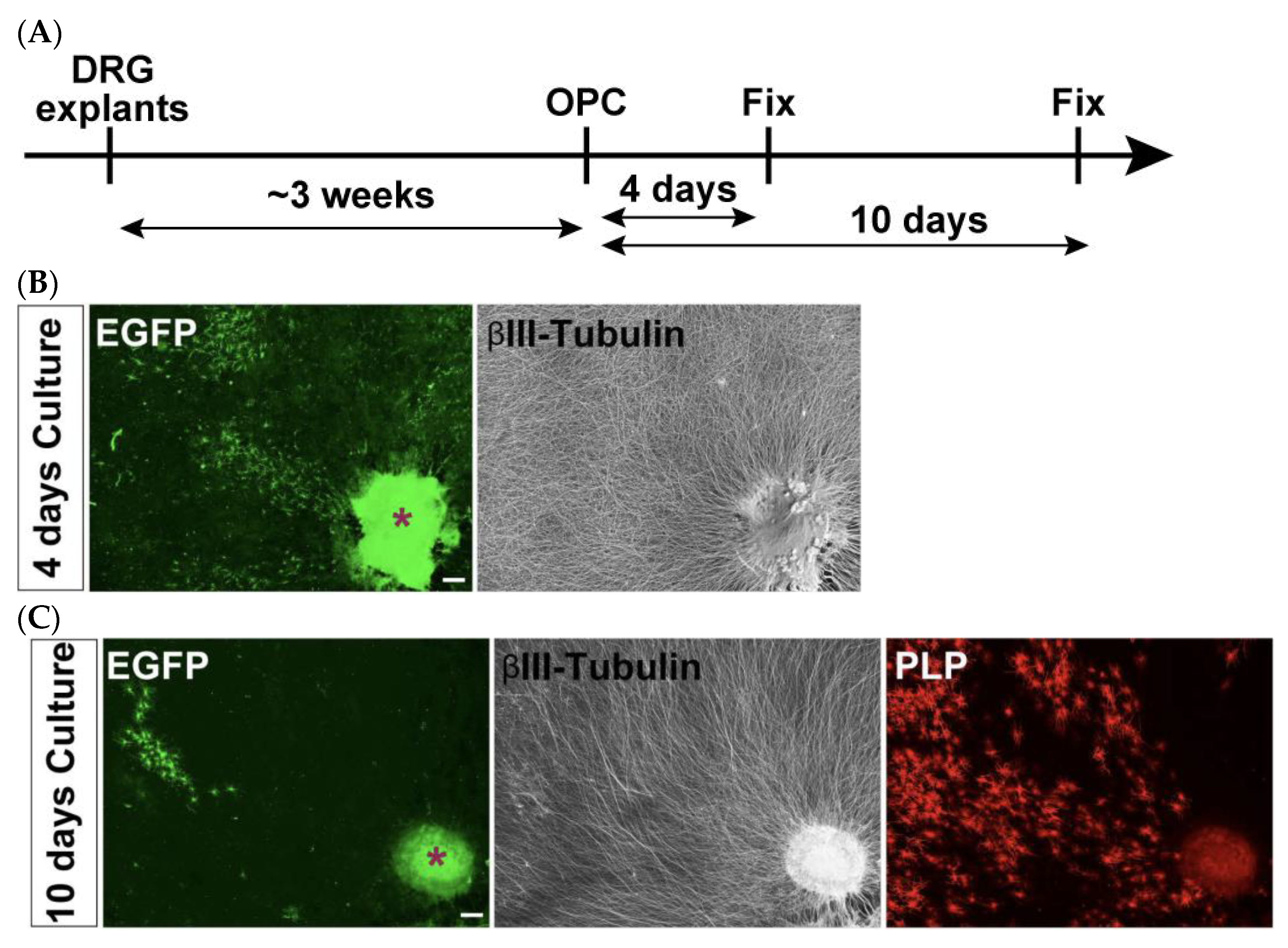
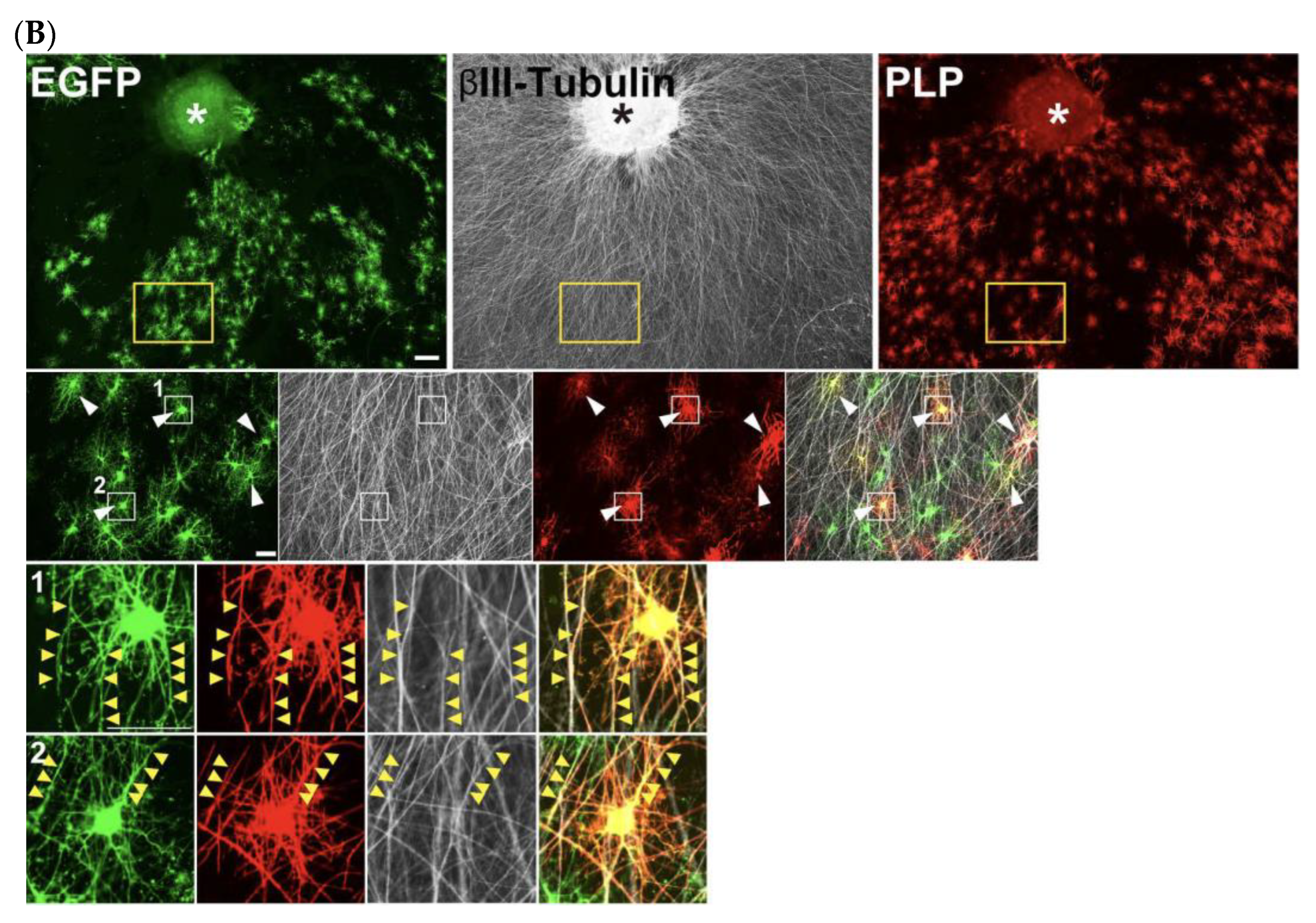
3.5. DRG Explants and Transfected OPCs Co-Culture (See Figure 4)
- Collect embryonic day 13 (E13.5) embryos from a time pregnant female mouse and collect DRGs from cervical and thoracic regions of the spinal cord.
- Plate 3–4 explants onto a 13 mm cover glass coated with PLO/MG-GFR in DRG medium (Table 3). Replace half of the medium every other day. During the first week, add FdU at a final concentration of 10 μM to eliminate proliferating cells, such as glial cells.
- Allow neuronal axons to extend radially. Axons are typically ready for co-culture with OPCs after 3 weeks of culture.
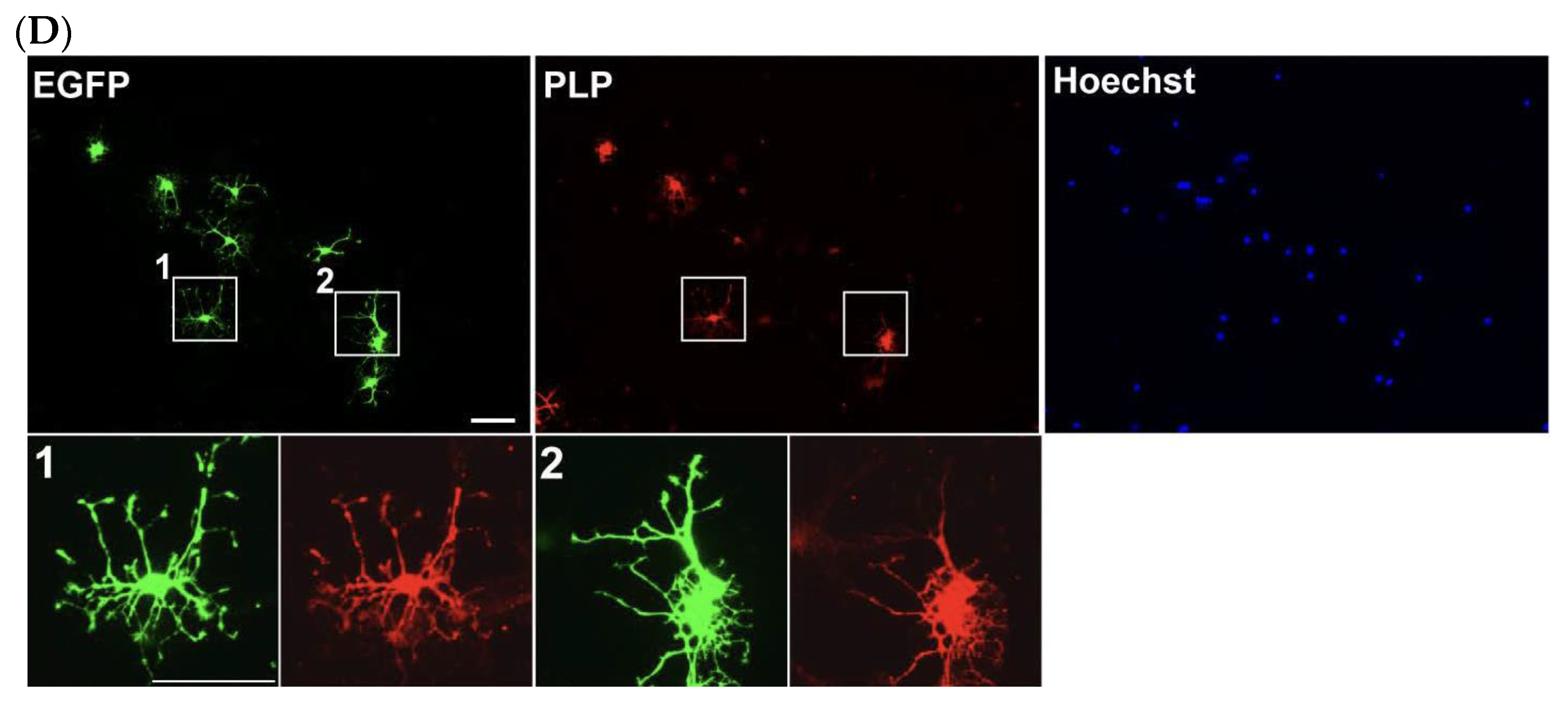
- For the neuron–glia co-culture, isolate OPCs as described above, transfect them with EGFP-expressing plasmids using electroporation, and plate the cells onto the established DRG explant culture.Note: For long-term co-culture, OPCs were electroporated with 5 μg of a PiggyBac Transposon vector encoding EGFP and 5 μg of a Super PiggyBac Transposase expression vector.Optional: Debris can be removed by passing the electroporated cell solution through a 40 μm mesh. However, a significant number of cells may be lost due to their retention on the mesh.
- Culture DRGs and OPCs in co-culture growth medium (Table 3) for 2 days.
- On day 2, replace the medium with co-culture differentiation medium (Table 3). Continue replacing half of the medium every other day until analysis.
3.6. Immunocytochemical Staining and Dead Cell Labeling
3.6.1. Dead Cell Labeling
- Prepare 1000× Dead Cell Makeup stock solution according to the product manual.
- Dilute the 1000× stock solution with the culture medium to prepare a 1× working solution.
- Replace the culture medium with the 1× Dead Cell Makeup working solution and incubate in a CO2 incubator for 15 min at 37 °C.
- Rinse the cells twice with PBS and proceed to the following immunostaining steps.
3.6.2. Immunocytochemical Staining
- Fix the cells with 4% PFA solution for 20 min at room temperature.
- Wash the cells 3 times with PBS and permeabilize cells with PBS containing 0.5% Triton X-100 (PBT) for 10 min at room temperature.
- Proceed to a blocking step by incubating PBT containing 10% normal donkey serum (blocking buffer) for 1 h.
- Remove the blocking buffer and apply first antibodies against targets such as GFP, PLP, and βIII-tubulin. Incubate overnight at 4 °C.
- Wash 3 times with PBS and apply fluorophore-conjugated secondary antibodies. Incubate the mixture for 2 h at room temperature.
- Wash 3 times with PBS and mount the coverslips for microscopic observation.
4. Expected Results
4.1. Efficiency of OPC Isolation and Their Viability
4.2. Gene Delivery into Isolated OPCs with an Electroporation Strategy
4.3. DRG Neuron–OPC Co-Culture for Neuron–Glia Interaction
5. Discussion and Conclusions
6. Troubleshooting
- Low cell recovery after tissue dissociation:This often results from excessive pipetting force when trying to obtain a single-cell suspension. Other contributing factors include the generation of bubbles and the use of pipette tips with sharp edges. To address this, gentle pipetting is recommended, and fire-polished glass pipettes should be used to minimize shear stress and mechanical damage.
- Clogging of the MS column:Failure to properly resuspend the cell pellet after centrifugation can lead to column clogging. Ensure the suspension is homogeneous with no visible clumps before loading onto the MS column.
- Low transfection efficiency during electroporation:A common cause is the presence of air bubbles in the electroporation cuvette. Use a glass pipette to slowly and carefully apply the cell suspension to avoid introducing bubbles. If bubbles are present, they should be removed either by gently tapping the cuvette or by using a glass pipette to aspirate them.
- High cell mortality after electroporation:Cells should be promptly transferred to culture medium after electroporation to improve viability. Moreover, plasmid DNA should be purified using endotoxin-free kits to minimize toxicity.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Simons, M.; Gibson, E.M.; Nave, K.-A. Oligodendrocytes: Myelination, Plasticity, and Axonal Support. Cold Spring Harb. Perspect. Biol. 2024, 16, a041359. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes Promote Neuronal Survival and Axonal Length by Distinct Intracellular Mechanisms: A Novel Role for Oligodendrocyte-Derived Glial Cell Line-Derived Neurotrophic Factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [PubMed]
- Pajevic, S.; Plenz, D.; Basser, P.J.; Fields, R.D. Oligodendrocyte-Mediated Myelin Plasticity and Its Role in Neural Synchronization. eLife 2023, 12, e81982. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Saglam, A.; Zuchero, J.B.; Buch, V.P. Translating Molecular Approaches to Oligodendrocyte-Mediated Neurological Circuit Modulation. Brain Sci. 2024, 14, 648. [Google Scholar] [CrossRef]
- Kedia, S.; Simons, M. Oligodendrocytes in Alzheimer’s Disease Pathophysiology. Nat. Neurosci. 2025, 28, 446–456. [Google Scholar] [CrossRef]
- Dehestani, M.; Kozareva, V.; Blauwendraat, C.; Fraenkel, E.; Gasser, T.; Bansal, V. Transcriptomic Changes in Oligodendrocytes and Precursor Cells Associate with Clinical Outcomes of Parkinson’s Disease. Mol. Brain 2024, 17, 56. [Google Scholar] [CrossRef]
- Zhu, B.; Park, J.-M.; Coffey, S.R.; Russo, A.; Hsu, I.-U.; Wang, J.; Su, C.; Chang, R.; Lam, T.T.; Gopal, P.P.; et al. Single-Cell Transcriptomic and Proteomic Analysis of Parkinson’s Disease Brains. Sci. Transl. Med. 2024, 16, eabo1997. [Google Scholar] [CrossRef]
- Takahashi, N.; Sakurai, T.; Davis, K.L.; Buxbaum, J.D. Linking Oligodendrocyte and Myelin Dysfunction to Neurocircuitry Abnormalities in Schizophrenia. Prog. Neurobiol. 2011, 93, 13–24. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, Y.; Liu, Z.; Geng, Q.; Chen, Z.; Zhang, Y. Alterations of Myelin Morphology and Oligodendrocyte Development in Early Stage of Alzheimer’s Disease Mouse Model. Neurosci. Lett. 2017, 642, 102–106. [Google Scholar] [CrossRef]
- Kagawa, T.; Ikenaka, K.; Inoue, Y.; Kuriyama, S.; Tsujii, T.; Nakao, J.; Nakajima, K.; Aruga, J.; Okano, H.; Mikoshiba, K. Glial Cell Degeneration and Hypomyelination Caused by Overexpression of Myelin Proteolipid Protein Gene. Neuron 1994, 13, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Massari, C.M.; Dues, D.J.; Bergsma, A.; Sipple, K.; Frye, M.; Williams, E.T.; Moore, D.J. Neuropathology in an α-Synuclein Preformed Fibril Mouse Model Occurs Independent of the Parkinson’s Disease-Linked Lysosomal ATP13A2 Protein. Neurobiol. Dis. 2024, 202, 106701. [Google Scholar] [CrossRef]
- Yu, G.; Su, Y.; Guo, C.; Yi, C.; Yu, B.; Chen, H.; Cui, Y.; Wang, X.; Wang, Y.; Chen, X.; et al. Pathological Oligodendrocyte Precursor Cells Revealed in Human Schizophrenic Brains and Trigger Schizophrenia-like Behaviors and Synaptic Defects in Genetic Animal Model. Mol. Psychiatry 2022, 27, 5154–5166. [Google Scholar] [CrossRef]
- O’Meara, R.W.; Ryan, S.D.; Colognato, H.; Kothary, R. Derivation of Enriched Oligodendrocyte Cultures and Oligodendrocyte/Neuron Myelinating Co-Cultures from Post-Natal Murine Tissues. J. Vis. Exp. 2011, 54, 3324. [Google Scholar] [CrossRef]
- Emery, B.; Dugas, J.C. Purification of Oligodendrocyte Lineage Cells from Mouse Cortices by Immunopanning. Cold Spring Harb. Protoc. 2013, 2013, pdb.prot073973. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.-L.; Cui, Q.-L.; Schambach, A.; Kim, K.-P.; Bachelin, C.; et al. Rapid and Efficient Generation of Oligodendrocytes from Human Induced Pluripotent Stem Cells Using Transcription Factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef] [PubMed]
- Krueger, W.H.; Madison, D.L.; Pfeiffer, S.E. Transient Transfection of Oligodendrocyte Progenitors by Electroporation. Neurochem. Res. 1998, 23, 421–426. [Google Scholar] [CrossRef]
- Xu, H.; Dzhashiashvili, Y.; Shah, A.; Kunjamma, R.B.; Weng, Y.; Elbaz, B.; Fei, Q.; Jones, J.S.; Li, Y.I.; Zhuang, X.; et al. m6A mRNA Methylation Is Essential for Oligodendrocyte Maturation and CNS Myelination. Neuron 2020, 105, 293–309.e5. [Google Scholar] [CrossRef]
- Powell, S.K.; Khan, N.; Parker, C.L.; Samulski, R.J.; Matsushima, G.; Gray, S.J.; McCown, T.J. Characterization of a Novel Adeno-Associated Viral Vector with Preferential Oligodendrocyte Tropism. Gene Ther. 2016, 23, 807–814. [Google Scholar] [CrossRef]
- Peckham, H.M.; Ferner, A.H.; Giuffrida, L.; Murray, S.S.; Xiao, J. Production and Use of Lentivirus to Selectively Transduce Primary Oligodendrocyte Precursor Cells for In Vitro Myelination Assays. J. Vis. Exp. 2015, 95, 52179. [Google Scholar] [CrossRef]
- Pringle, N.P.; Mudhar, H.S.; Collarini, E.J.; Richardson, W.D. PDGF Receptors in the Rat CNS: During Late Neurogenesis, PDGF Alpha-Receptor Expression Appears to Be Restricted to Glial Cells of the Oligodendrocyte Lineage. Development 1992, 115, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Hart, I.K.; Richardson, W.D.; Heldin, C.-H.; Westermark, B.; Raff, M.C. PDGF Receptors on Cells of the Oligodendrocyte-Type-2 Astrocyte (O-2A) Cell Lineage. Development 1989, 105, 595–603. [Google Scholar] [CrossRef]
- Ishino, Y.; Shimizu, S.; Tohyama, M.; Miyata, S. Coactivator-associated Arginine Methyltransferase 1 Controls Oligodendrocyte Differentiation in the Corpus Callosum during Early Brain Development. Dev. Neurobiol. 2022, 82, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Konola, J.T.; Wekerle, H.; Lees, M.B. Monoclonal Antibodies against Myelin Proteolipid Protein: Identification and Characterization of Two Major Determinants. J. Neurochem. 1991, 57, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Leites, E.P.; Morais, V.A. Protocol for the Isolation and Culture of Microglia, Astrocytes, and Neurons from the Same Mouse Brain. STAR Protoc. 2024, 5, 102804. [Google Scholar] [CrossRef]
- Dincman, T.A.; Beare, J.E.; Ohri, S.S.; Whittemore, S.R. Isolation of Cortical Mouse Oligodendrocyte Precursor Cells. J. Neurosci. Methods 2012, 209, 219–226. [Google Scholar] [CrossRef]
- Jensen, C.J.; Demol, F.; Bauwens, R.; Kooijman, R.; Massie, A.; Villers, A.; Ris, L.; De Keyser, J. Astrocytic Β2 Adrenergic Receptor Gene Deletion Affects Memory in Aged Mice. PLoS ONE 2016, 11, e0164721. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Lee, D.R.; Kim, H.-S.; Yoo, J.-E.; Jung, S.J.; Lim, B.Y.; Jang, J.; Kang, H.-C.; You, S.; Hwang, D.-Y.; et al. Highly Pure and Expandable PSA-NCAM-Positive Neural Precursors from Human ESC and iPSC-Derived Neural Rosettes. PLoS ONE 2012, 7, e39715. [Google Scholar] [CrossRef]
- Weider, M.; Starost, L.J.; Groll, K.; Küspert, M.; Sock, E.; Wedel, M.; Fröb, F.; Schmitt, C.; Baroti, T.; Hartwig, A.C.; et al. Nfat/Calcineurin Signaling Promotes Oligodendrocyte Differentiation and Myelination by Transcription Factor Network Tuning. Nat. Commun. 2018, 9, 899. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, Z.; Wang, Y.; Li, Y.; Lü, H.; Fu, S.; Hang, Q.; Lu, P.-H. Oligodendrocyte–Spinal Cord Explant Co-Culture: An in Vitro Model for the Study of Myelination. Brain Res. 2010, 1309, 9–18. [Google Scholar] [CrossRef]
- Pang, Y.; Zheng, B.; Kimberly, S.L.; Cai, Z.; Rhodes, P.G.; Lin, R.C.S. Neuron-oligodendrocyte Myelination Co-culture Derived from Embryonic Rat Spinal Cord and Cerebral Cortex. Brain Behav. 2012, 2, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wong, A.W.; Willingham, M.M.; Van Den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-Derived Neurotrophic Factor Promotes Central Nervous System Myelination via a Direct Effect upon Oligodendrocytes. Neurosignals 2010, 18, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K. piggyBac Transposon. Microbiol. Spectr. 2015, 3, MDNA3-0028-2014. [Google Scholar] [CrossRef] [PubMed]
- Cary, L.C.; Goebel, M.; Corsaro, B.G.; Wang, H.-G.; Rosen, E.; Fraser, M.J. Transposon Mutagenesis of Baculoviruses: Analysis of Trichoplusia Ni Transposon IFP2 Insertions within the FP-Locus of Nuclear Polyhedrosis Viruses. Virology 1989, 172, 156–169. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight Control of Gene Expression in Mammalian Cells by Tetracycline-Responsive Promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Recipe |
|---|---|
| Enzyme Mix 1 | 12.5 μL Enzyme P and 487.5 μL Buffer X |
| Enzyme Mix 2 | 20 μL Buffer Y and 10 μL Enzyme A |
| Name | Composition |
|---|---|
| OPC proliferation medium | DMEM/F12, B27 supplement, 10 ng/mL PDGF-AA, 10 ng/mL CNTF, 1 ng/mL NT3 |
| OPC differentiation medium | DMEM/F12, B27 supplement, 10 ng/mL CNTF, 30 ng/mL T3, 5 μg/mL NAC |
| Name | Composition |
|---|---|
| DRG medium | Neurobasal medium, B27supplement, 10 ng/mL NGF |
| Co-culture growth medium | Neurobasal medium:DMEM/F12 = 1:1 B27 supplement, N2 supplement, 10 ng/mL NGF, 10 ng/mL PDGF-AA, 10 ng/mL CNTF, 1 ng/mL NT3 |
| Co-culture differentiation medium | Neurobasal medium:DMEM/F12 = 1:1 B27 supplement, N2 supplement, 10 ng/mL NGF, 10 ng/mL CNTF, 30 ng/mL T3, 5 μg/mL NAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishino, Y.; Shimizu, S.; Miyata, S. An Efficient Electroporation Protocol Supporting In Vitro Studies of Oligodendrocyte Biology. Methods Protoc. 2025, 8, 64. https://doi.org/10.3390/mps8030064
Ishino Y, Shimizu S, Miyata S. An Efficient Electroporation Protocol Supporting In Vitro Studies of Oligodendrocyte Biology. Methods and Protocols. 2025; 8(3):64. https://doi.org/10.3390/mps8030064
Chicago/Turabian StyleIshino, Yugo, Shoko Shimizu, and Shingo Miyata. 2025. "An Efficient Electroporation Protocol Supporting In Vitro Studies of Oligodendrocyte Biology" Methods and Protocols 8, no. 3: 64. https://doi.org/10.3390/mps8030064
APA StyleIshino, Y., Shimizu, S., & Miyata, S. (2025). An Efficient Electroporation Protocol Supporting In Vitro Studies of Oligodendrocyte Biology. Methods and Protocols, 8(3), 64. https://doi.org/10.3390/mps8030064