
A Refined Approach to Isolate Interneurons for High-Validity Epigenetic Studies in Human Brain Tissue

 ,
,  ,
,
Abstract
1. Introduction
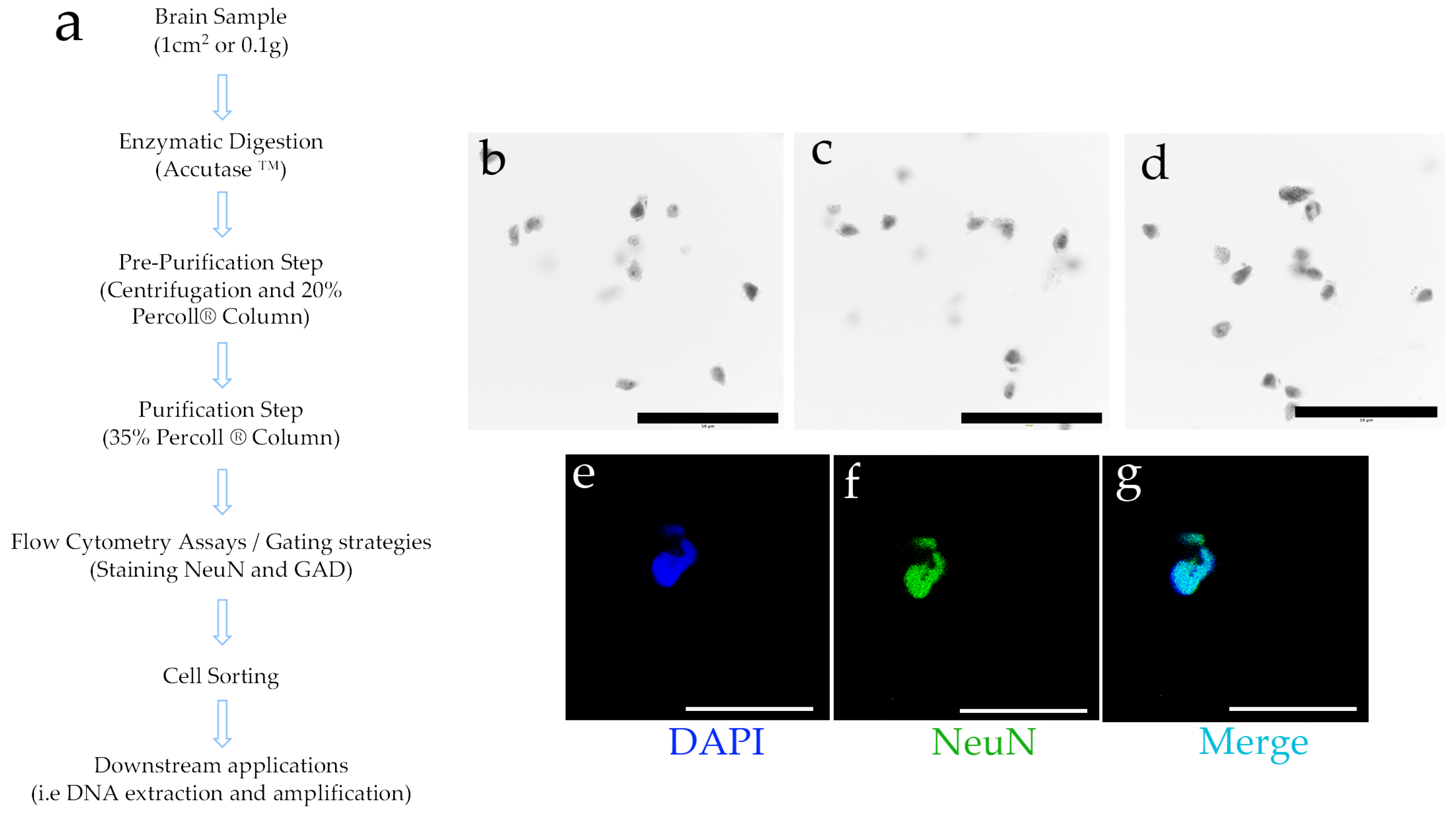
2. Materials and Methods
2.1. Samples
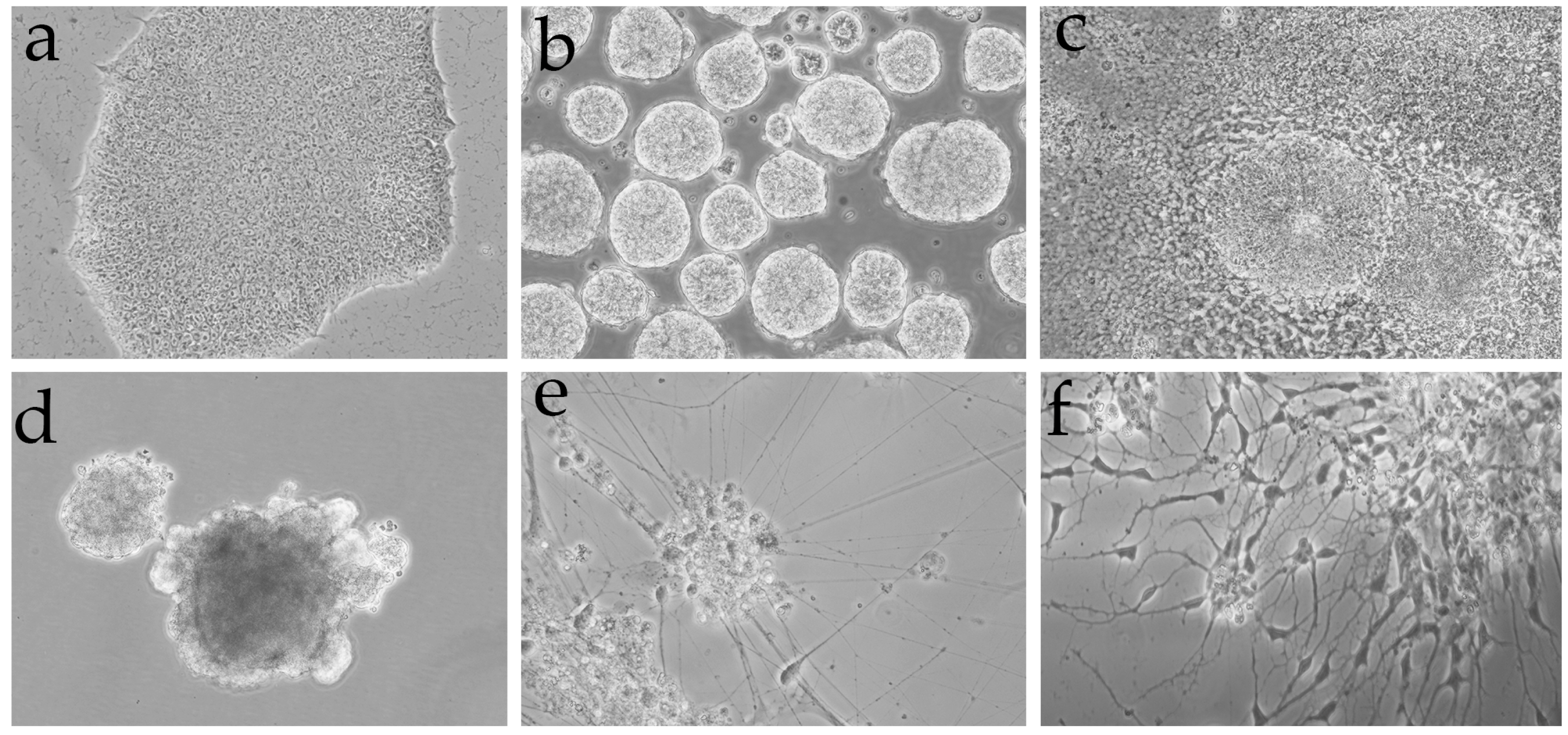
2.2. Cell Culture and Differentiation
2.3. Cell Separation
2.4. Antibodies
- -
- Anti-NeuN Alexa Fluor™ 488 conjugated (Millipore, Cat. MAB377X, Burlington, MA, USA); 1/50 for all experiments.
- -
- Anti-NeuN (clone 60) (Millipore, Cat. MAB377, Burlington, MA, USA); 1/350 for all experiments.
- -
- Anti-GAD 65/67 (Sigma, Cat. G5163, St. Louis, MO, USA); 1/1000 for experiments involving tissues and 1/250 for experiments involving cells.
- -
- Anti-FOXG1 (Abcam, Cat. Ab18259, Cambridge, UK); 1/500 for all experiments.
- -
- Antio-TTF1 (Abcam, Cat. Ab76013, Cambridge, UK); 1/200 for all experiments.
- -
- Anti-Class III β Tubulin (MO, USA, Sigma, Cat. T8660); 1/500 for all experiments.
- -
- Goat anti-rabbit (Alexa Fluor™ 488, ThermoFisher, Cat. A11034, Waltham, MA, USA); 1/500 for confocal microscopy experiments and 1/1000 for flow cytometry assays.
- -
- Goat anti-mouse (Alexa Fluor™ 647, ThermoFisher, Cat. A21236, Waltham, MA, USA); 1/500 for confocal microscopy experiments and 1/1000 for flow cytometry assays.
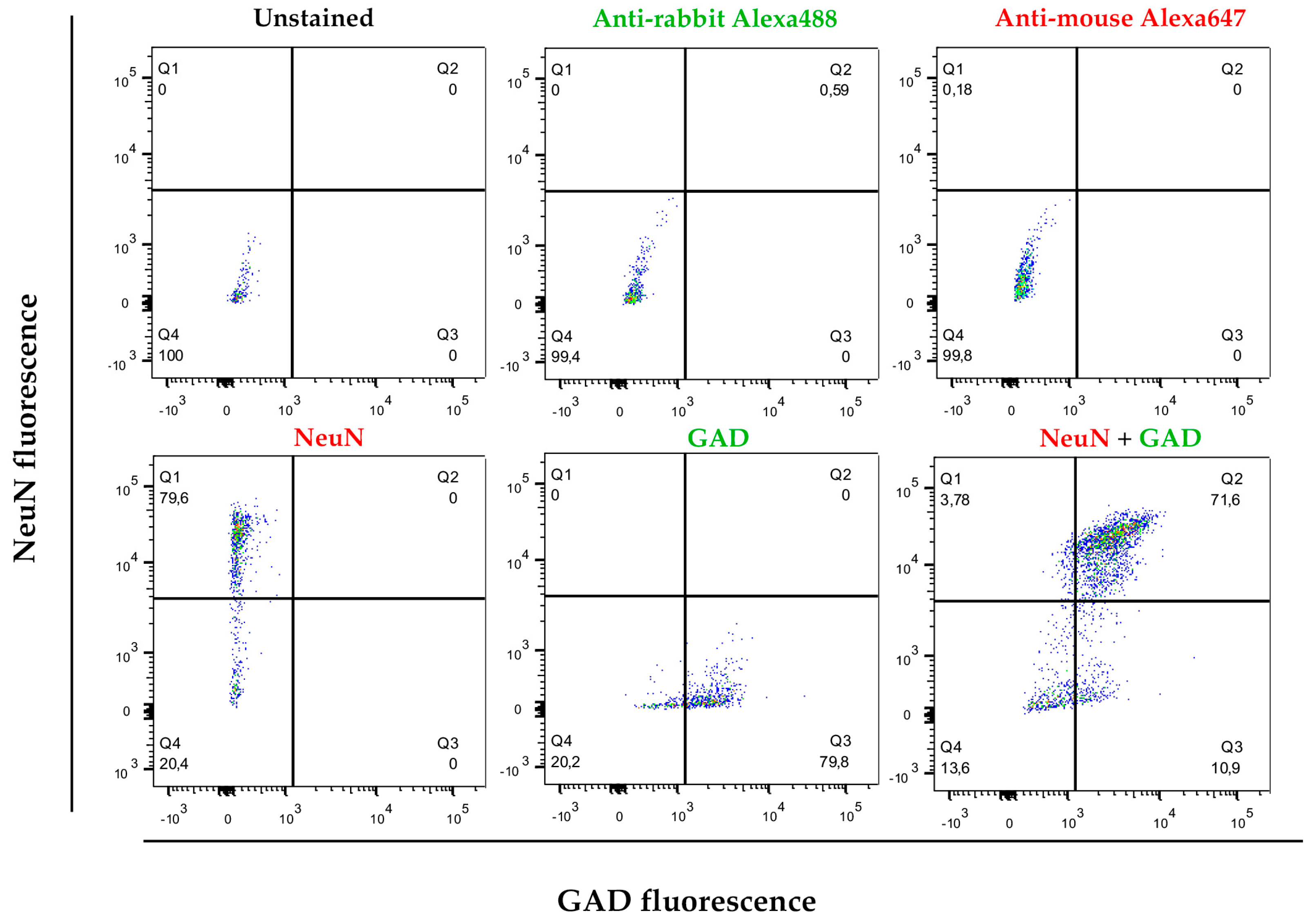
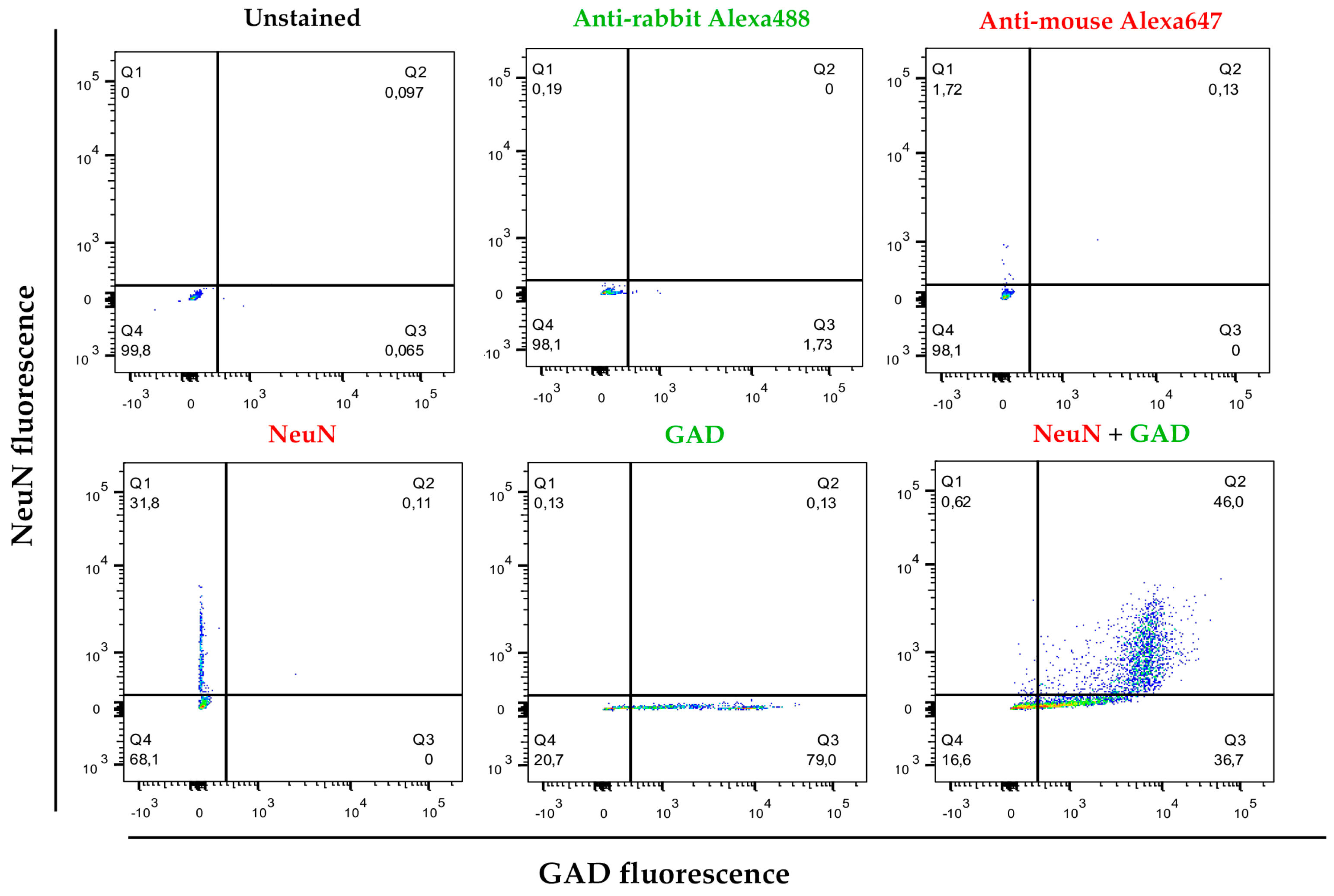
2.5. Cytometry Analysis and Cell Sorting
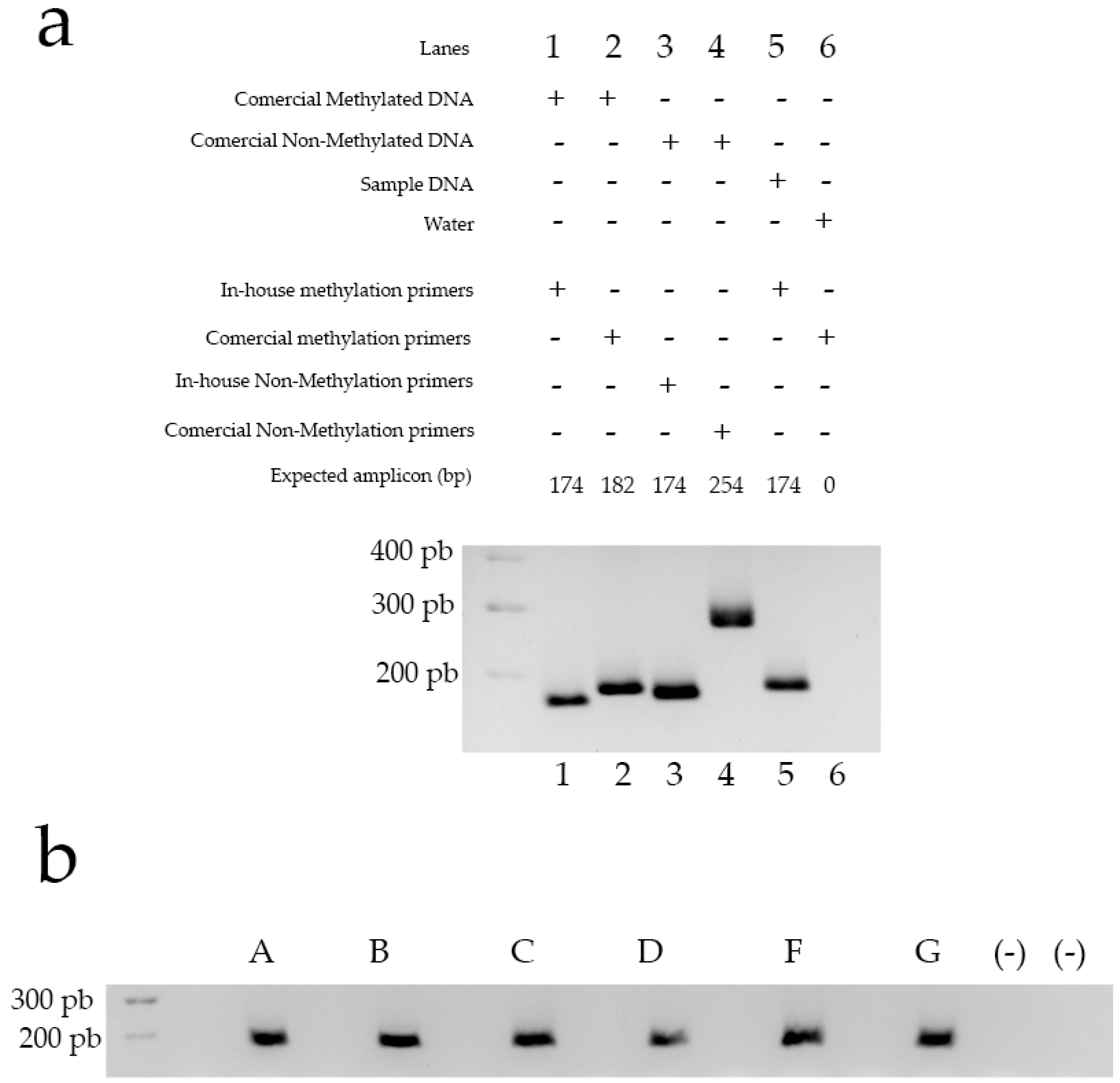
2.6. DNA Extraction, Modification and Amplification
2.7. Primer Validation and Controls
2.8. Confocal Microscopy
2.9. Statistical Analysis
3. Results
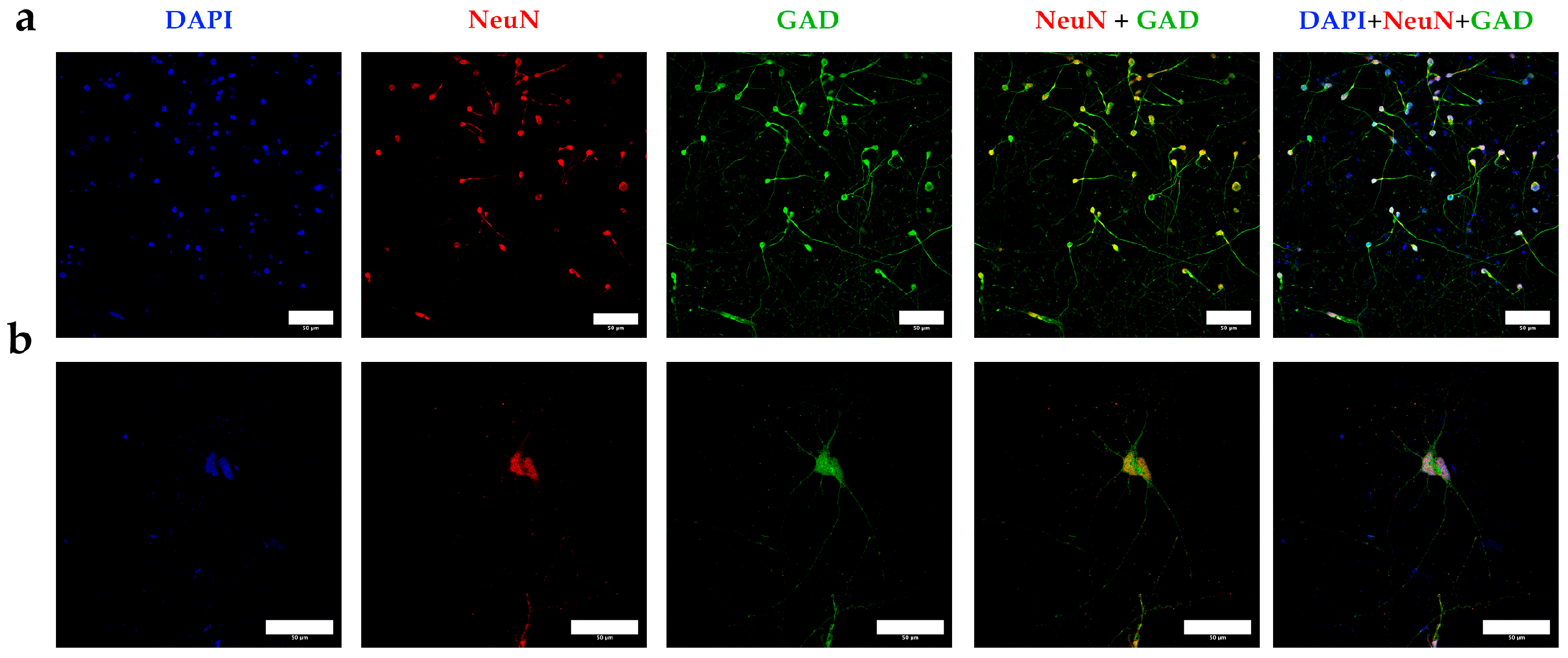
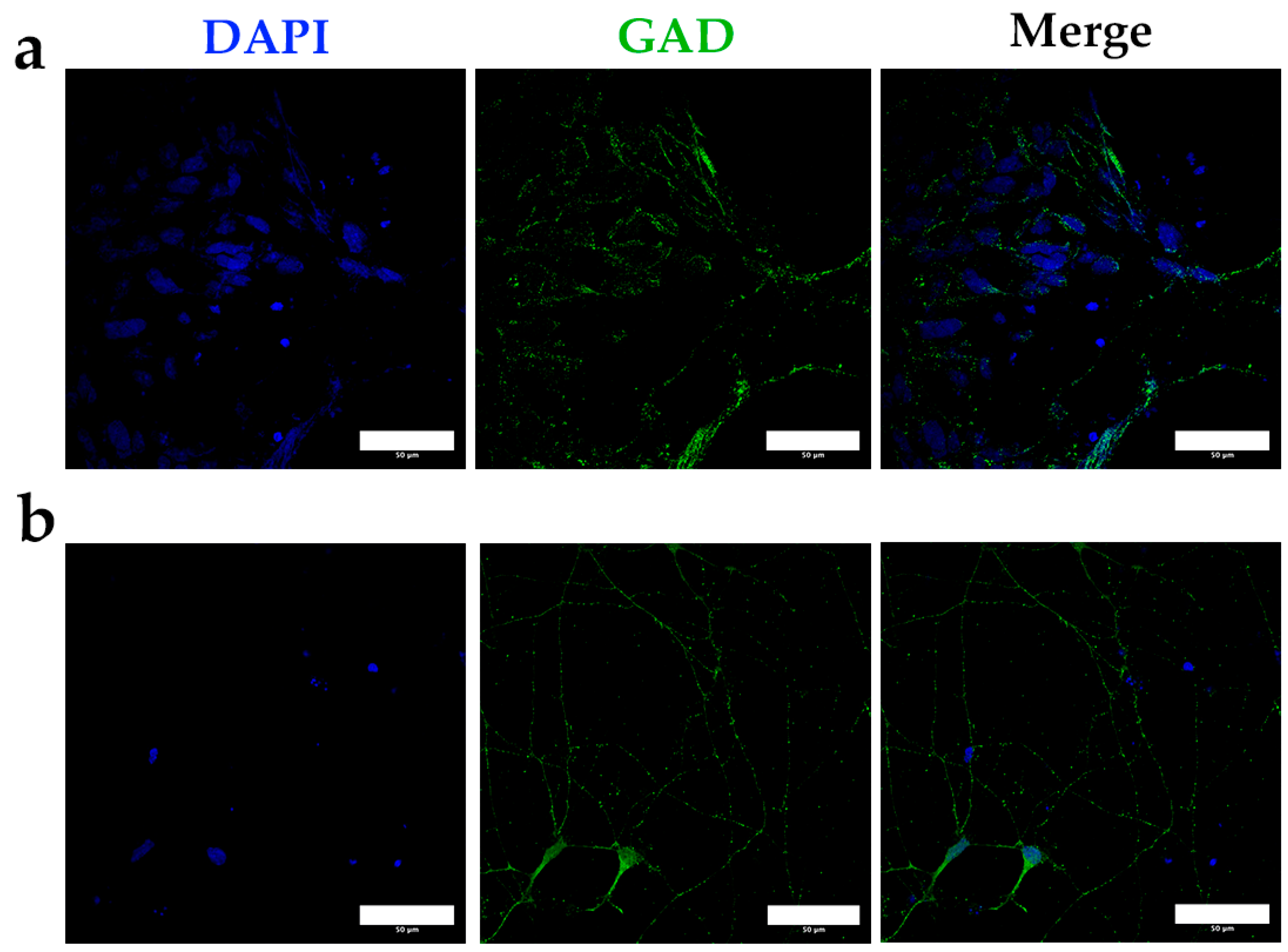
3.1. Enzymatic Digestion and Differential Density Gradients Allow Adequate Separation of Brain Tissue Cells
3.2. Cell Separation by Using Differential Densities May Help to Purify and Concentrate Cell Populations of Interest
3.3. The Obtained Genetic Material Can Be Used for Downstream Applications, in This Case, Methylation-Specific PCR
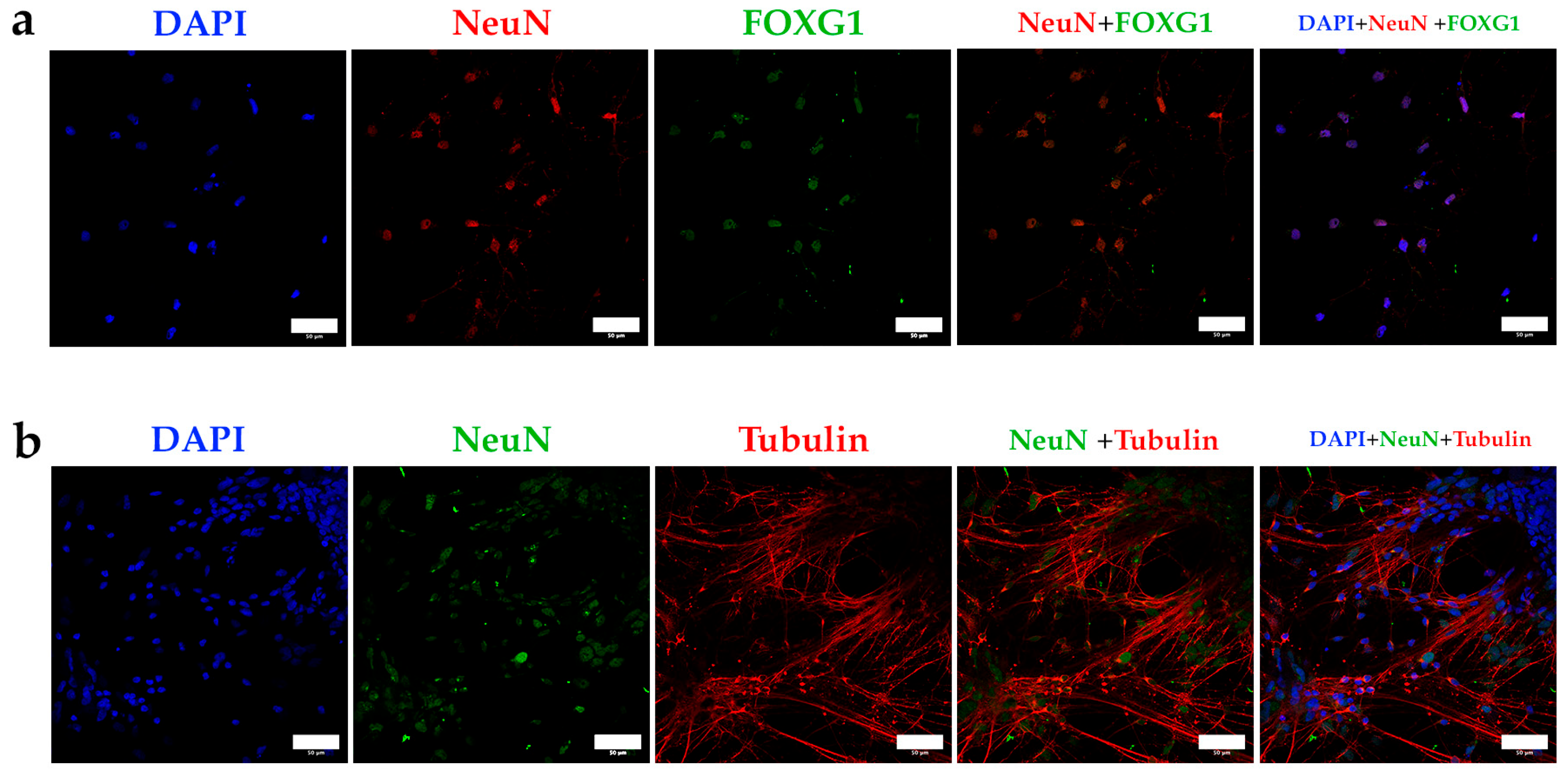
3.4. The Criteria Developed in This Protocol Can Be Contrasted by Using Interneurons Differentiated from Adult Induced-Pluripotent Stem Cells (iPSC)
3.5. The Markers Applied to the Separation of Populations in Brain Tissue Samples Separate a Discrete Population in Cultures of Interneurons Obtained by Reprogramming
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheah, S.-Y.; Lawford, B.R.; Young, R.M.; Morris, C.P.; Voisey, J. mRNA Expression and DNA Methylation Analysis of Serotonin Receptor 2A (HTR2A) in the Human Schizophrenic Brain. Genes 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; Palazzo, M.C.; Benatti, B.; Grancini, B.; Pucci, M.; Di Francesco, A.; Camuri, G.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; et al. Regulation of Gene Transcription in Bipolar Disorders: Role of DNA Methylation in the Relationship between Prodynorphin and Brain Derived Neurotrophic Factor. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Miura, I.; Kunii, Y.; Hino, M.; Hoshino, H.; Matsumoto, J.; Kanno-Nozaki, K.; Horikoshi, S.; Kaneko, H.; Bundo, M.; Iwamoto, K.; et al. DNA Methylation of ANKK1 and Response to Aripiprazole in Patients with Acute Schizophrenia: A Preliminary Study. J. Psychiatr. Res. 2018, 100, 84–87. [Google Scholar] [CrossRef]
- Lam, D.; Ancelin, M.-L.; Ritchie, K.; Freak-Poli, R.; Saffery, R.; Ryan, J. Genotype-Dependent Associations between Serotonin Transporter Gene (SLC6A4) DNA Methylation and Late-Life Depression. BMC Psychiatry 2018, 18, 282. [Google Scholar] [CrossRef]
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.E.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 Alternative Transcripts and DNA Methylation in Human Prefrontal Cortex and Hippocampus in Brain Development, Schizophrenia. Mol. Psychiatry 2018, 23, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA Methylation Associates the Cystine-Glutamate Antiporter SLC7A11 with Risk of Parkinson’s Disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef]
- Cariaga-Martinez, A.; Alelú-Paz, R. False Data, Positive Results in Neurobiology: Moving beyond the Epigenetics of Blood and Saliva Samples in Mental Disorders. J. Negat. Results Biomed. 2016, 15, 21. [Google Scholar] [CrossRef]
- Nabil Fikri, R.M.; Norlelawati, A.T.; Nour El-Huda, A.R.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Nor Zamzila, A. Reelin (RELN) DNA Methylation in the Peripheral Blood of Schizophrenia. J. Psychiatr. Res. 2017, 88, 28–37. [Google Scholar] [CrossRef]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-Control Meta-Analysis of Blood DNA Methylation and Autism Spectrum Disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef]
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA Methylation Signatures for Autism Spectrum Disorder Genomic Risk Loci: 16p11.2 Deletions and CHD8 Variants. Clin. Epigenet. 2019, 11, 103. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.; Qin, H.; Tubbs, J.D.; Li, M.; Qiao, C.; Lin, J.; Li, Q.; Fan, F.; Gou, M.; et al. Genome-Wide DNA Methylation Analysis of Peripheral Blood Cells Derived from Patients with First-Episode Schizophrenia in the Chinese Han Population. Mol. Psychiatry 2021, 26, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, J.; Qing, L.; Li, J.; Yang, H.; Ji, A.; Yan, M.; Hu, L.; Nie, S. DNA Methylation Analysis of the NR3C1 Gene in Patients with Schizophrenia. J. Mol. Neurosci. 2020, 70, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.-B.; Potash, J.B.; Feinberg, A.P. DNA Methylation Signatures within the Human Brain. Am. J. Hum. Genet. 2007, 81, 1304–1315. [Google Scholar] [CrossRef]
- Kato, T.; Iwamoto, K. Comprehensive DNA Methylation and Hydroxymethylation Analysis in the Human Brain and Its Implication in Mental Disorders. Neuropharmacology 2014, 80, 133–139. [Google Scholar] [CrossRef]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical Parvalbumin Interneurons and Cognitive Dysfunction in Schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Rapanelli, M.; Frick, L.R.; Pittenger, C. The Role of Interneurons in Autism and Tourette Syndrome. Trends Neurosci. 2017, 40, 397–407. [Google Scholar] [CrossRef]
- Ferguson, B.R.; Gao, W.-J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef]
- Dienel, S.J.; Lewis, D.A. Alterations in Cortical Interneurons and Cognitive Function in Schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Anderson, K.M.; Collins, M.A.; Chin, R.; Ge, T.; Rosenberg, M.D.; Holmes, A.J. Transcriptional and Imaging-Genetic Association of Cortical Interneurons, Brain Function, and Schizophrenia Risk. Nat. Commun. 2020, 11, 2889. [Google Scholar] [CrossRef]
- Markram, H.; Toledo-Rodriguez, M.; Wang, Y.; Gupta, A.; Silberberg, G.; Wu, C. Interneurons of the Neocortical Inhibitory System. Nat. Rev. Neurosci. 2004, 5, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Dracheva, S.; Elhakem, S.L.; McGurk, S.R.; Davis, K.L.; Haroutunian, V. GAD67 and GAD65 mRNA and Protein Expression in Cerebrocortical Regions of Elderly Patients with Schizophrenia. J. Neurosci. Res. 2004, 76, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Karolewicz, B.; Maciag, D.; O’Dwyer, G.; Stockmeier, C.A.; Feyissa, A.M.; Rajkowska, G. Reduced Level of Glutamic Acid Decarboxylase-67 kDa in the Prefrontal Cortex in Major Depression. Int. J. Neuropsychopharmacol. 2010, 13, 411–420. [Google Scholar] [CrossRef]
- Fish, K.N.; Sweet, R.A.; Lewis, D.A. Differential Distribution of Proteins Regulating GABA Synthesis and Reuptake in Axon Boutons of Subpopulations of Cortical Interneurons. Cereb. Cortex 2011, 21, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Le Magueresse, C.; Monyer, H. GABAergic Interneurons Shape the Functional Maturation of the Cortex. Neuron 2013, 77, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-G.; Yao, R.; Monnell, T.; Cho, J.-H.; Vasudevan, A.; Koh, A.; Peeyush, K.T.; Moon, M.; Datta, D.; Bolshakov, V.Y.; et al. Efficient Specification of Interneurons from Human Pluripotent Stem Cells by Dorsoventral and Rostrocaudal Modulation. Stem Cells 2014, 32, 1789–1804. [Google Scholar] [CrossRef]
- Le, T.N.; Zhou, Q.-P.; Cobos, I.; Zhang, S.; Zagozewski, J.; Japoni, S.; Vriend, J.; Parkinson, T.; Du, G.; Rubenstein, J.L.; et al. GABAergic Interneuron Differentiation in the Basal Forebrain Is Mediated through Direct Regulation of Glutamic Acid Decarboxylase Isoforms by Dlx Homeobox Transcription Factors. J. Neurosci. 2017, 37, 8816–8829. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Sotuyo, N.; Tischfield, D.J.; Anderson, S.A. Generation of Cerebral Cortical GABAergic Interneurons from Pluripotent Stem Cells. Stem Cells 2020, 38, 1375–1386. [Google Scholar] [CrossRef]
- Akbarian, S. Epigenetic Mechanisms in Schizophrenia. Dialogues Clin. Neurosci. 2014, 16, 405–417. [Google Scholar] [CrossRef]
- Murphy, B.C.; O’Reilly, R.L.; Singh, S.M. Site-Specific Cytosine Methylation in S-COMT Promoter in 31 Brain Regions with Implications for Studies Involving Schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 133B, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleky, H.M.; Cheng, K.-H.; Faraone, S.V.; Wilcox, M.; Glatt, S.J.; Gao, F.; Smith, C.L.; Shafa, R.; Aeali, B.; Carnevale, J.; et al. Hypomethylation of MB-COMT Promoter Is a Major Risk Factor for Schizophrenia and Bipolar Disorder. Hum. Mol. Genet. 2006, 15, 3132–3145. [Google Scholar] [CrossRef]
- Alelú-Paz, R.; Carmona, F.J.; Sanchez-Mut, J.V.; Cariaga-Martínez, A.; González-Corpas, A.; Ashour, N.; Orea, M.J.; Escanilla, A.; Monje, A.; Guerrero Márquez, C.; et al. Epigenetics in Schizophrenia: A Pilot Study of Global DNA Methylation in Different Brain Regions Associated with Higher Cognitive Functions. Front. Psychol. 2016, 7, 1496. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.; Smith, R.G.; Jeffries, A.; Hannon, E.; Davies, M.N.; Schalkwyk, L.; Mill, J.; Weedon, M.; Lunnon, K. Regional Differences in Mitochondrial DNA Methylation in Human Post-Mortem Brain Tissue. Clin. Epigenet. 2017, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Fachim, H.A.; Srisawat, U.; Dalton, C.F.; Reynolds, G.P. Parvalbumin Promoter Hypermethylation in Postmortem Brain in Schizophrenia. Epigenomics 2018, 10, 519–524. [Google Scholar] [CrossRef]
- Smith, A.R.; Smith, R.G.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Kalaria, R.N.; Sloan, C.; Robinson, A.C.; Mill, J.; Lunnon, K. A Cross-Brain Regions Study of ANK1 DNA Methylation in Different Neurodegenerative Diseases. Neurobiol. Aging 2019, 74, 70–76. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-Wide DNA Methylation Profiling Identifies Convergent Molecular Signatures Associated with Idiopathic and Syndromic Autism in Post-Mortem Human Brain Tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef]
- Kozlenkov, A.; Wang, M.; Roussos, P.; Rudchenko, S.; Barbu, M.; Bibikova, M.; Klotzle, B.; Dwork, A.J.; Zhang, B.; Hurd, Y.L.; et al. Substantial DNA Methylation Differences between Two Major Neuronal Subtypes in Human Brain. Nucleic Acids Res. 2016, 44, 2593–2612. [Google Scholar] [CrossRef]
- Mai, J.; Majtanik, M.; Paxinos, G. Atlas of the Human Brain, 3rd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Liu, Y.; Liu, H.; Sauvey, C.; Yao, L.; Zarnowska, E.D.; Zhang, S.-C. Directed Differentiation of Forebrain GABA Interneurons from Human Pluripotent Stem Cells. Nat. Protoc. 2013, 8, 1670–1679. [Google Scholar] [CrossRef]
- Gusel’nikova, V.V.; Korzhevskiy, D.E. NeuN As a Neuronal Nuclear Antigen and Neuron Differentiation Marker. Acta Naturae 2015, 7, 42–47. [Google Scholar] [CrossRef]
- Xuan, S.; Baptista, C.A.; Balas, G.; Tao, W.; Soares, V.C.; Lai, E. Winged Helix Transcription Factor BF-1 Is Essential for the Development of the Cerebral Hemispheres. Neuron 1995, 14, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.-S.; hAilín, D.Ó.; Vogel, T.; Hanashima, C. Transcription and Beyond: Delineating FOXG1 Function in Cortical Development and Disorders. Front. Cell. Neurosci. 2020, 14, 35. [Google Scholar] [CrossRef]
- Goulburn, A.L.; Stanley, E.G.; Elefanty, A.G.; Anderson, S.A. Generating GABAergic Cerebral Cortical Interneurons from Mouse and Human Embryonic Stem Cells. Stem Cell Res. 2012, 8, 416–426. [Google Scholar] [CrossRef]
- Sandberg, M.; Flandin, P.; Silberberg, S.; Su-Feher, L.; Price, J.D.; Hu, J.S.; Kim, C.; Visel, A.; Nord, A.S.; Rubenstein, J.L.R. Transcriptional Networks Controlled by NKX2-1 in the Development of Forebrain GABAergic Neurons. Neuron 2016, 91, 1260–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In Vitro Differentiation of Transplantable Neural Precursors from Human Embryonic Stem Cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Li, J.; Apontes, P.; Hurd, Y.L.; Byne, W.M.; Koonin, E.V.; Wegner, M.; Mukamel, E.A.; Dracheva, S. A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv. 2018, 4, eaau6190. [Google Scholar] [CrossRef]
- Douglas, L.N.; McGuire, A.B.; Manzardo, A.M.; Butler, M.G. High-resolution chromosome ideogram representation of recognized genes for bipolar disorder. Gene 2016, 586, 136–147. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saini, S.M.; Mancuso, S.G.; Mostaid, M.S.; Liu, C.; Pantelis, C.; Everall, I.P.; Bousman, C.A. Meta-analysis supports GWAS-implicated link between GRM3 and schizophrenia risk. Transl. Psychiatry 2017, 7, e1196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dogra, S.; Stansley, B.J.; Xiang, Z.; Qian, W.; Gogliotti, R.G.; Nicoletti, F.; Lindsley, C.W.; Niswender, C.M.; Joffe, M.E.; Conn, P.J. Activating mGlu3 Metabotropic Glutamate Receptors Rescues Schizophrenia-like Cognitive Deficits Through Metaplastic Adaptations Within the Hippocampus. Biol. Psychiatry 2021, 90, 385–398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, C.Y.; Xiao, X.; Zhang, Z.; Hu, Z.; Li, M. An alternative splicing hypothesis for neuropathology of schizophrenia: Evidence from studies on historical candidate genes and multi-omics data. Mol. Psychiatry 2022, 27, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Eschenlauer, A.; Aman, J.; Folsom, T.D.; Chekouo, T. Quantitative proteomics of dorsolateral prefrontal cortex reveals an early pattern of synaptic dysmaturation in children with idiopathic autism. Cereb. Cortex 2024, 34, 161–171. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maj, M. Understanding the Pathophysiology of Schizophrenia: Are We on the Wrong or on the Right Track? Schizophr. Res. 2011, 127, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, S.S.; De Boni, U. Pure Population of Viable Neurons from Rabbit Dorsal Root Ganglia, Using Gradients of Percoll. J. Neurobiol. 1983, 14, 195–206. [Google Scholar] [CrossRef]
- Strong, M.J.; Garruto, R.M. Isolation of Fetal Mouse Motor Neurons on Discontinuous Percoll Density Gradients. In Vitro Cell Dev. Biol. 1989, 25, 939–945. [Google Scholar] [CrossRef]
- Katzenell, S.; Cabrera, J.R.; North, B.J.; Leib, D.A. Isolation, Purification, and Culture of Primary Murine Sensory Neurons. Methods Mol. Biol. 2017, 1656, 229–251. [Google Scholar] [CrossRef]
- Bernocco, S.; Fondelli, C.; Matteoni, S.; Magnoni, L.; Gotta, S.; Terstappen, G.C.; Raggiaschi, R. Sequential Detergent Fractionation of Primary Neurons for Proteomics Studies. Proteomics 2008, 8, 930–938. [Google Scholar] [CrossRef]
- Sun, Y.; Ip, P.; Chakrabartty, A. Simple Elimination of Background Fluorescence in Formalin-Fixed Human Brain Tissue for Immunofluorescence Microscopy. J. Vis. Exp. 2017, 56188. [Google Scholar] [CrossRef]
- Pyon, W.S.; Gray, D.T.; Barnes, C.A. An Alternative to Dye-Based Approaches to Remove Background Autofluorescence From Primate Brain Tissue. Front. Neuroanat. 2019, 13, 73. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Sex | Age | Cause of Death | Postmortem Delay (h) |
|---|---|---|---|---|
| 1 | Male | 46 | Ischemia | 5 |
| 2 | Male | 54 | Myocardial infarct | 5 |
| 3 | Male | 67 | Ischemia | 4 |
| 4 | Male | 69 | Ischemia | 16 |
| 5 | Male | 79 | Ischemia | 2 |
| 8 | Male | 81 | Ischemia | 10 |
| Name | Sequence 5′-3′ | Tm (°C) |
|---|---|---|
| GRM3-MS | TTT AGT ATT TTC GGA TGG GC | 60.22 |
| GRM3-MAS | CGC GAC TCA ACT ACA CAC TAC | 60.14 |
| GRM3-NMS | TAG TTT AGT ATT TTT GGA TGG GT | 56.95 |
| GRM3-NMAS | CTC ACA ACT CAA CTA CAC ACT ACA | 58.5 |
| Lane | Sample DNA | Primer | Sequence | Expected Amplicon (bp) | Observed Amplicon (bp) |
|---|---|---|---|---|---|
| 1 | Methylated commercial DNA | In-house primer designed to detect methylated sites. | MS (TTT AGT ATT TTC GGA TGG GC) MAS (CGC GAC TCA ACT ACA CAC TAC) | 174 | 174 |
| 2 | Methylated comercial DNA | Comercial primer designed to detect methylated sites. | Unknown | 182 | 182 |
| 3 | Non-Methylated comercial DNA | In-house primer designed to detect non-methylated sites. | NMS (TAG TTT AGT ATT TTT GGA TGG GT) NMAS (CTC ACA ACT CAA CTA CAC ACT ACA) | 174 | 174 |
| 4 | Non-Methylated comercial DNA | Comercial primer designed to detect non-methylated sites. | Unknown | 254 | 254 |
| 5 | Modified DNA obtained from isolated cells | In-house primer designed to detect methylated sites. | MS (TTT AGT ATT TTC GGA TGG GC) MAS (CGC GAC TCA ACT ACA CAC TAC) | 174 | 174 |
| 6 | No-DNA Sample (Water) | Comercial primer designed to detect methylated sites. | Unknown | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cariaga-Martínez, A.; Gutierrez, K.J.; Regidor, I.; Del Álamo, M.; Saiz-Ruiz, J.; Alelú-Paz, R. A Refined Approach to Isolate Interneurons for High-Validity Epigenetic Studies in Human Brain Tissue. Methods Protoc. 2025, 8, 61. https://doi.org/10.3390/mps8030061
Cariaga-Martínez A, Gutierrez KJ, Regidor I, Del Álamo M, Saiz-Ruiz J, Alelú-Paz R. A Refined Approach to Isolate Interneurons for High-Validity Epigenetic Studies in Human Brain Tissue. Methods and Protocols. 2025; 8(3):61. https://doi.org/10.3390/mps8030061
Chicago/Turabian StyleCariaga-Martínez, Ariel, Kilian Jesús Gutierrez, Ignacio Regidor, Marta Del Álamo, Jerónimo Saiz-Ruiz, and Raúl Alelú-Paz. 2025. "A Refined Approach to Isolate Interneurons for High-Validity Epigenetic Studies in Human Brain Tissue" Methods and Protocols 8, no. 3: 61. https://doi.org/10.3390/mps8030061
APA StyleCariaga-Martínez, A., Gutierrez, K. J., Regidor, I., Del Álamo, M., Saiz-Ruiz, J., & Alelú-Paz, R. (2025). A Refined Approach to Isolate Interneurons for High-Validity Epigenetic Studies in Human Brain Tissue. Methods and Protocols, 8(3), 61. https://doi.org/10.3390/mps8030061