


Specific Aspects of SELEX Protocol: Different Approaches for ssDNA Generation

 ,
, 
Abstract
1. Introduction
2. Experimental Design
2.1. Design of Study
2.2. Materials
- Chloroform (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.:210100);
- Phenol (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.:200121);
- Ammonium acetate (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.:010299);
- Isoamyl alcohol (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.: 010131);
- Ethylenediaminetetraacetic acid (EDTA); (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.: 600047);
- Urea (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.: 120427);
- Magnesium acetate tetrahydrate (Lenreaktiv Ltd., Saint Petersburg, Russia, Cat.no.: 120062);
- A 40% acrylamide/N,N’-methylenebisacrylamide 19:1 solution (Bio-Rad Laboratories, Hercules, CA, USA, Cat.no.: 1610145);
- Formamide (Khimmed Ltd., Saint Petersburg, Russia, Cat.no.: BP227-500);
- Sodium dodecyl sulfate and TBE ×10 buffer, pH 8.3 (Dia-M Ltd., Saint Petersburg, Russia, Cat.no.: LC-10112.1 and Cat.no.: 3774.0250);
- Ethidium bromide 10 mg/mL (Helicon Ltd., Saint Petersburg, Russia, Cat.no.: EtBr-10). Considering the high toxicity of ethidium bromide, alternative reagents (SYBR Green I/II, GelRed/GelGreen, Methylene Blue, Novel Juice) can be used instead;
- DNA marker Step50 plus and 4 × buffer for applying DNA to the gel “BiK” (both from Biolabmix Ltd., Novosibirsk, Russia, Cat.no.: S-8055), GeneRuler 50bp DNA Ladder Thermo Fisher Scientific, Waltham, MA, USA, Cat.no.: SM0372);
- Lambda phage exonuclease and 10× exonuclease reaction buffer (both from New England Biolabs, Frankfurt, Germany, Cat.no.: M0262S);
- Ethyl alcohol 96%;
- PCR-grade water (Biolabmix Ltd., Novosibirsk, Russia, Cat.no.: SP010-05),
- PCR master mix «BioMaster HS-Taq PCR x2» (Biolabmix Ltd., Novosibirsk, Russia, Cat.no.: MH010-200). Mix included 100 mM Tris-HCl, pH 8.5, 100 mM KCl, 0.4 mM of each dNTP, 4 mM MgCl2, 0.06 units of activity/μlTaq DNA polymerase and 0.2% Tween 20;
- Synthetic oligonucleotides (Syntol Ltd., Moscow, Russia). Sequences presented in Table 1. Phosphorylated (5′-end) reverse primers from various suppliers were used for comparative analysis.
2.3. Equipment
- DNA amplifier “BIS” M111-02-48 (Chaldin, Novosibirsk, Russia);
- MicroCL 17R Centrifuge (Thermo Fisher Scientific, Waltham, MA, USA);
- iBright™ FL1000 Imaging System (Thermo Fisher Scientific, Waltham, MA, USA);
- Vertical Electrophoresis Chamber VE-10-v2 (Helicon, Moscow, Russia);
- PowerPacTM Basic Power Supply (Bio-Rad Laboratories, Hercules, NY, USA);
- Thermo shaker TS-100 (BioSan, Riga, Latvia);
- NanoPhotometer N50 spectrophotometer (Implen, Munchen, Germany).
3. Procedure
3.1. Polymerase Chain Reaction
3.2. dsDNA Extraction from the Reaction Mixtures
3.3. Exonuclease Digestion
3.4. Polyacrylamide Gel Electroforesis (PAGE)
3.5. Denaturing PAGE (dPAGE)
3.6. ssDNA Extraction from Polyacrylamide Gel
3.7. Statistical Analysis
4. Protocol Setup
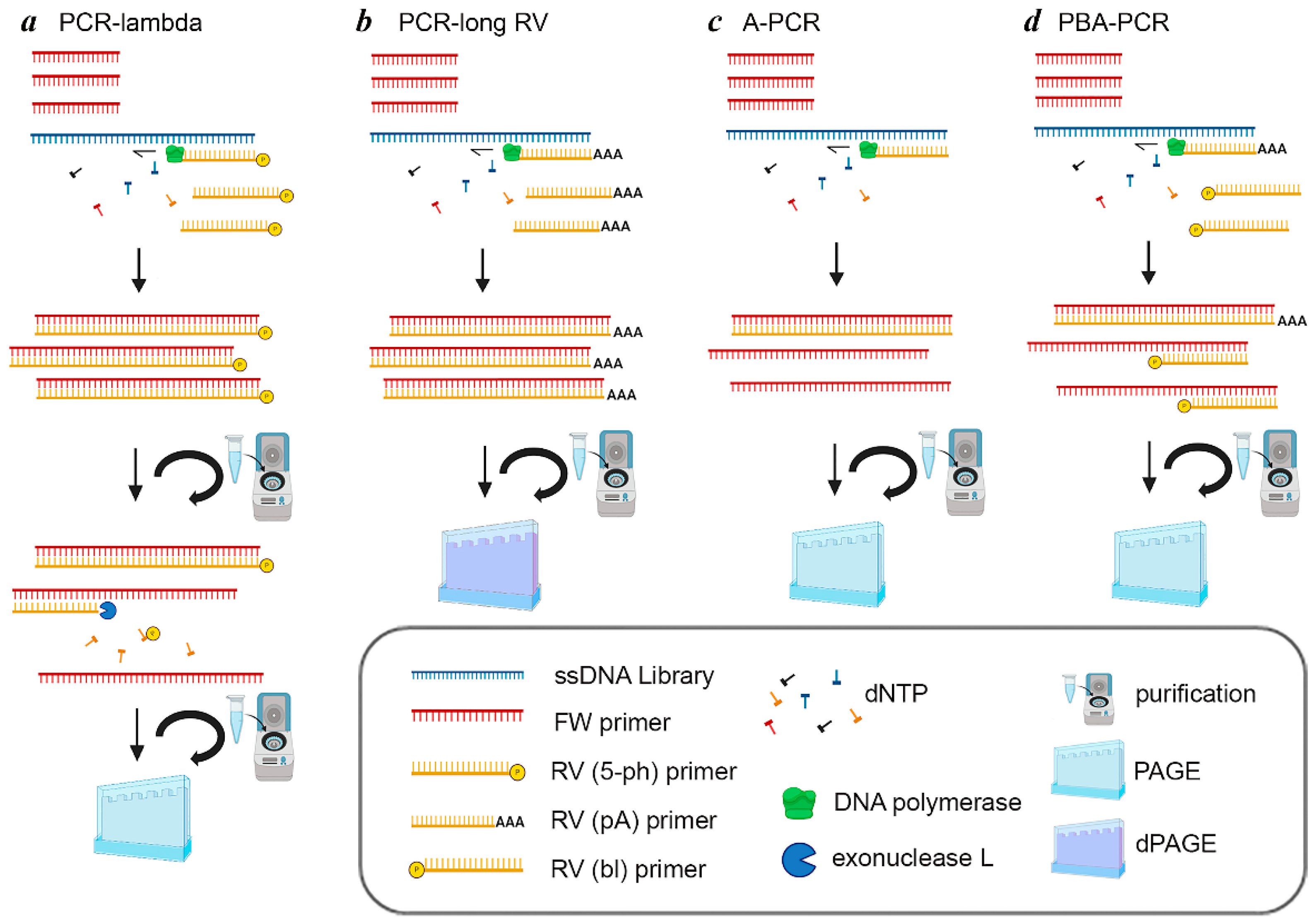
4.1. Specific Aspects of PCR-Lambda
4.2. Specific Aspects of PCR-Long RV
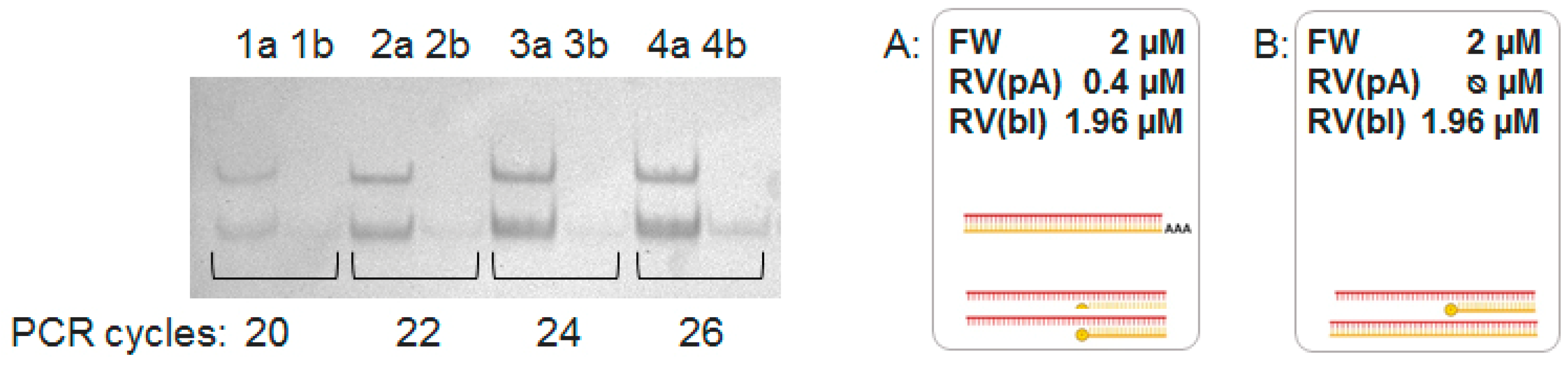
4.3. Specific Aspects of A-PCR and PBA-PCR
5. Results and Discussion
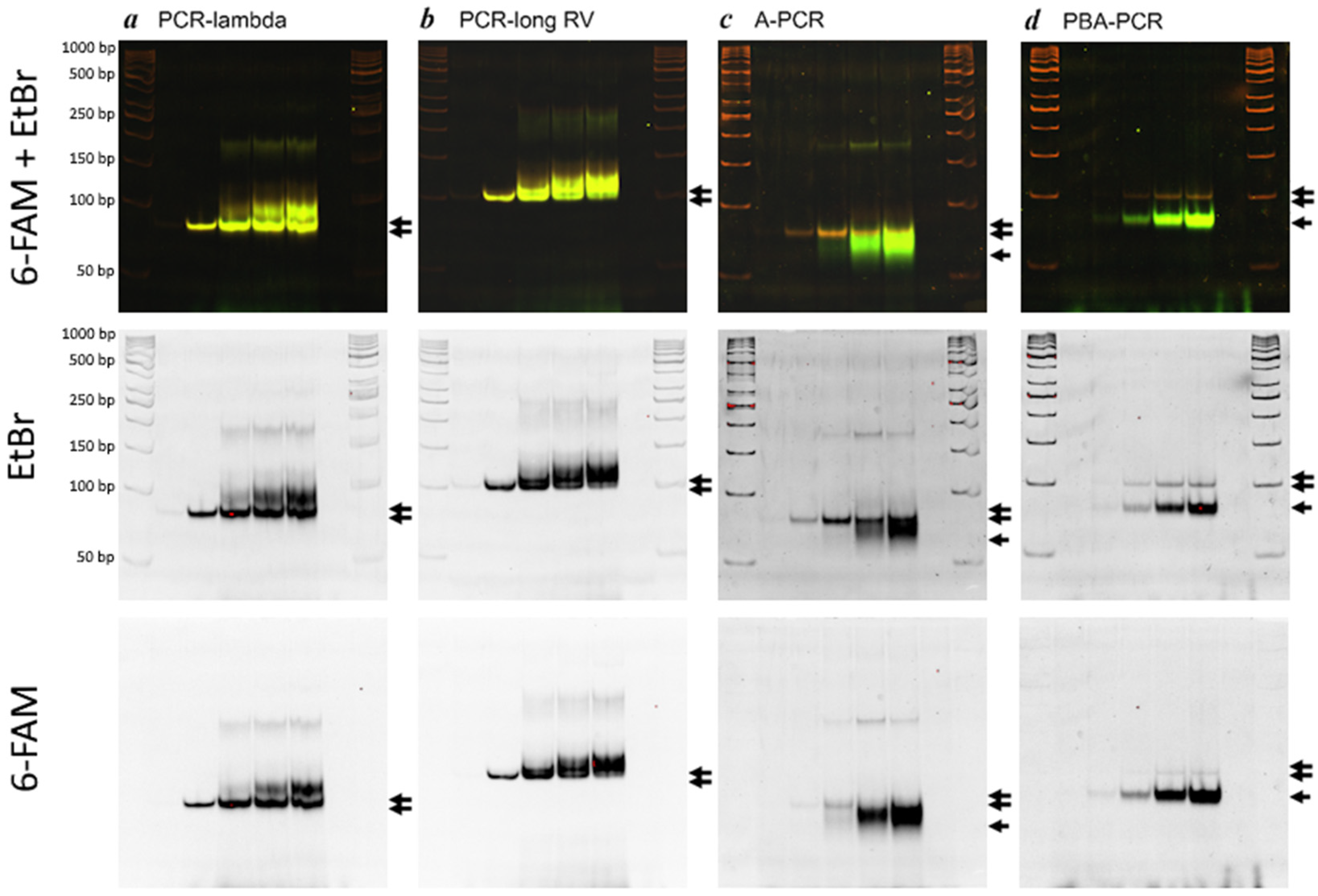
5.1. Evalution of PCR Specificity
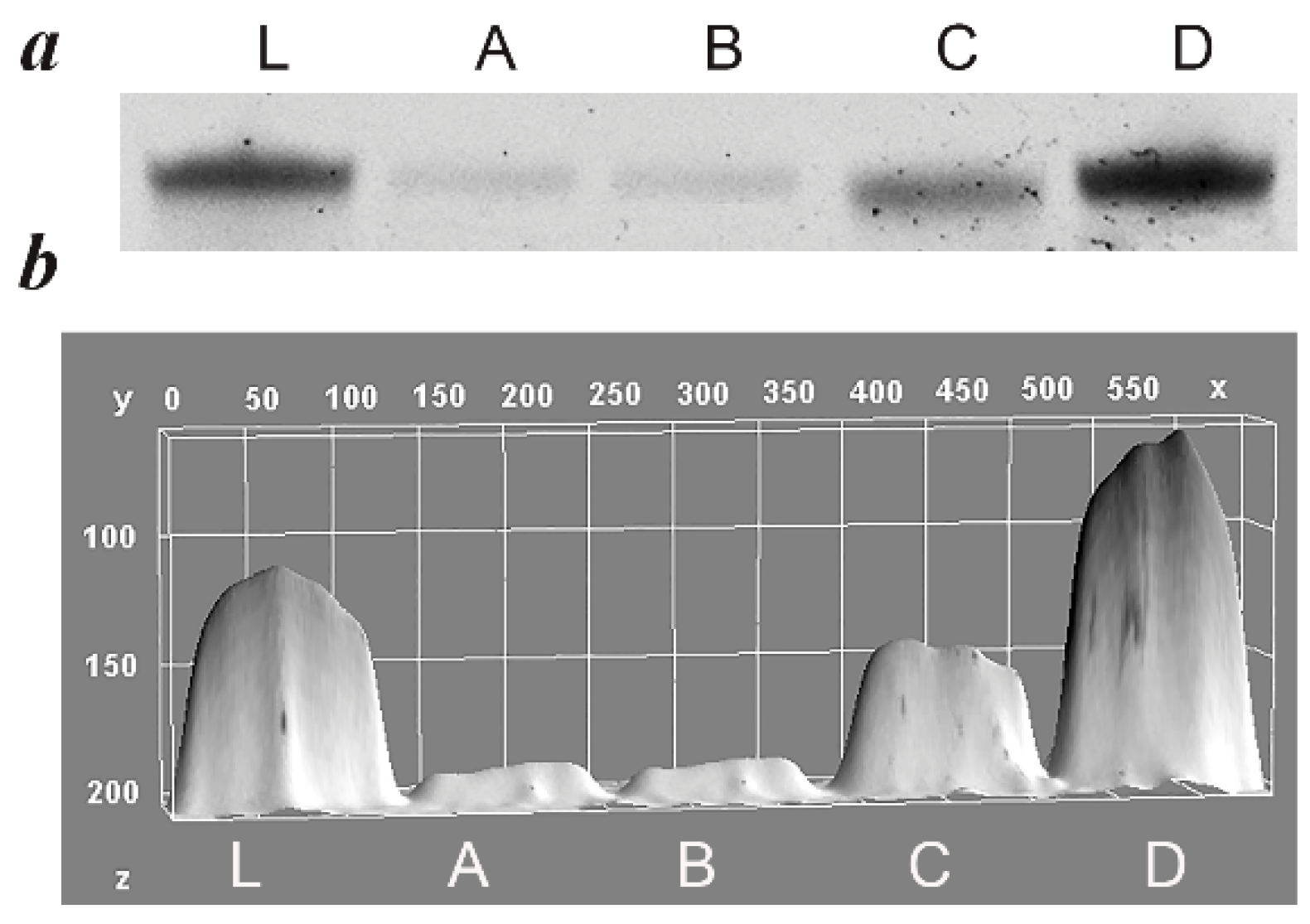
5.2. Evaluation of Efficacy of ssDNA Generation
5.3. Reproducibility and Feasibility of Methods
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SELEX | Systematic evolution of ligands by exponential enrichment |
| ssDNA | Single-stranded deoxyribonucleic acid |
| dsDNA | Double-stranded deoxyribonucleic acid |
| PCR | Polymerase chain reaction |
| PAGE | Polyacrylamide gel electrophoresis |
| dPAGE | Denaturing urea polyacrylamide gel electrophoresis |
| TBE buffer | Tris-borate-EDTA buffer |
| A-PCR | Asymmetric polymerase chain reaction |
| PBA-PCR | Primer-blocked asymmetric polymerase chain reaction |
References
- Röthlisberger, P.; Hollenstein, M. Aptamer Chemistry. Adv. Drug Deliv. Rev. 2018, 134, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, D.; Zeng, Z.; Huang, L.; Lin, X.; Hong, S. Aptamer-Based Probes for Cancer Diagnostics and Treatment. Life 2022, 12, 1937. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Kohlberger, M.; Gadermaier, G. SELEX: Critical Factors and Optimization Strategies for Successful Aptamer Selection. Biotechnol. Appl. Biochem. 2022, 69, 1771–1792. [Google Scholar] [CrossRef]
- Williams, K.P.; Bartel, D.P. PCR Product with Strands of Unequal Length. Nucleic Acids Res. 1995, 23, 4220–4221. [Google Scholar] [CrossRef]
- Paul, A.; Avci-Adali, M.; Ziemer, G.; Wendel, H.P. Streptavidin-Coated Magnetic Beads for DNA Strand Separation Implicate a Multitude of Problems During Cell-SELEX. Oligonucleotides 2009, 19, 243–254. [Google Scholar] [CrossRef]
- Wilson, R. Preparation of Single-Stranded DNA from PCR Products with Streptavidin Magnetic Beads. Nucleic Acid Ther. 2011, 21, 437–440. [Google Scholar] [CrossRef]
- Kilili, G.K.; Tilton, L.; Karbiwnyk, C.M. [Letter to the Editor] NaOH Concentration and Streptavidin Bead Type Are Key Factors for Optimal DNA Aptamer Strand Separation and Isolation. Biotechniques 2016, 61, 114–116. [Google Scholar] [CrossRef]
- Gowland, R.; Gowers, D.M. A Lambda-Exonuclease SELEX Method for Generating Aptamers to Bacterial Targets. In DNA Manipulation and Analysis; Springer: New York, NY, USA, 2023; pp. 145–161. [Google Scholar]
- Avci-Adali, M.; Paul, A.; Wilhelm, N.; Ziemer, G.; Wendel, H.P. Upgrading SELEX Technology by Using Lambda Exonuclease Digestion for Single-Stranded DNA Generation. Molecules 2009, 15, 1–11. [Google Scholar] [CrossRef]
- Damase, T.R.; Ellington, A.D.; Allen, P.B. Purification of Single-Stranded DNA by Co-Polymerization with Arylamide and Electrophoresis. Biotechniques 2017, 62, 275–282. [Google Scholar] [CrossRef]
- Coombes, P.E.; Dickman, M.J. Optimisation of Denaturing Ion Pair Reversed Phase HPLC for the Purification of SsDNA in SELEX. J. Chromatogr. A 2024, 1719, 464699. [Google Scholar] [CrossRef] [PubMed]
- Gyllensten, U.B.; Erlich, H.A. Generation of Single-Stranded DNA by the Polymerase Chain Reaction and Its Application to Direct Sequencing of the HLA-DQA Locus. Proc. Natl. Acad. Sci. USA 1988, 85, 7652–7656. [Google Scholar] [CrossRef] [PubMed]
- Yunusov, D.; So, M.; Shayan, S.; Okhonin, V.; Musheev, M.U.; Berezovski, M.V.; Krylov, S.N. Kinetic Capillary Electrophoresis-Based Affinity Screening of Aptamer Clones. Anal. Chim. Acta 2009, 631, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tabarzad, M.; Kazemi, B.; Vahidi, H.; Aboofazeli, R.; Shahhosseini, S.; Nafissi-Varcheh, N. Challenges to Design and Develop of DNA Aptamers for Protein Targets. I. Optimization of Asymmetric PCR for Generation of a Single Stranded DNA Library. Iran. J. Pharm. Res. IJPR 2014, 13, 133–141. [Google Scholar] [PubMed]
- He, C.-Z.; Zhang, K.-H.; Wang, T.; Wan, Q.-S.; Hu, P.-P.; Hu, M.-D.; Huang, D.-Q.; Lv, N.-H. Single-Primer-Limited Amplification: A Method to Generate Random Single-Stranded DNA Sub-Library for Aptamer Selection. Anal. Biochem. 2013, 440, 63–70. [Google Scholar] [CrossRef]
- Yeoh, T.S.; Anna, A.; Tang, T.-H.; Citartan, M. Development of an Optimization Pipeline of Asymmetric PCR towards the Generation of DNA Aptamers: A Guide for Beginners. World J. Microbiol. Biotechnol. 2022, 38, 31. [Google Scholar] [CrossRef]
- Tolnai, Z.; Harkai, Á.; Szeitner, Z.; Scholz, É.N.; Percze, K.; Gyurkovics, A.; Mészáros, T. A Simple Modification Increases Specificity and Efficiency of Asymmetric PCR. Anal. Chim. Acta 2019, 1047, 225–230. [Google Scholar] [CrossRef]
- Nehdi, A.; Samman, N.; Aguilar-Sánchez, V.; Farah, A.; Yurdusev, E.; Boudjelal, M.; Perreault, J. Novel Strategies to Optimize the Amplification of Single-Stranded DNA. Front. Bioeng. Biotechnol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Marimuthu, C.; Tang, T.-H.; Tominaga, J.; Tan, S.-C.; Gopinath, S.C.B. Single-Stranded DNA (SsDNA) Production in DNA Aptamer Generation. Analyst 2012, 137, 1307. [Google Scholar] [CrossRef]
- Svobodová, M.; Pinto, A.; Nadal, P.; O’ Sullivan, C.K. Comparison of Different Methods for Generation of Single-Stranded DNA for SELEX Processes. Anal. Bioanal. Chem. 2012, 404, 835–842. [Google Scholar] [CrossRef]
- Liang, C.; Li, D.; Zhang, G.; Li, H.; Shao, N.; Liang, Z.; Zhang, L.; Lu, A.; Zhang, G. Comparison of the Methods for Generating Single-Stranded DNA in SELEX. Analyst 2015, 140, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Percze, K.; Harkai, Á.; Mészáros, T. A Cost-Effective Approach for Single-Stranded DNA Amplification Using Primer-Blocked Asymmetric PCR. Curr. Protoc. 2024, 4, e1125. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Isolation of DNA Fragments from Polyacrylamide Gels by the Crush and Soak Method. Cold Spring Harb. Protoc. 2019, 2019, pdb.prot100479. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure 5′→3′ | Length, nts |
|---|---|---|
| ssDNA library * | TAGGGAAGAGAAGGACATATGAT-(N)30-TTGACTAGTACATGACCACTTGA | 76 |
| FW | TAGGGAAGAGAAGGACATATGAT | 23 |
| FW(FAM) | [6-FAM]-TAGGGAAGAGAAGGACATATGAT | 23 |
| RV | TCAAGTGGTCATGTACTAGTCAA | 23 |
| RV(5′-ph) | phosphate-TCAAGTGGTCATGTACTAGTCAA | 23 |
| RV-poly(dA) | AAAAAAAAAAAAAAAAAAAA(spacer18)TCAAGTGGTCATGTACTAGTCAA | 43 |
| RV(bl) | TCAAGTGGTCATGTACTAGTCAA-phosphate | 23 |
| Primers | PCR-lambda | PCR-long RV | A-PCR | PBA-PCR |
|---|---|---|---|---|
| FW/FW(FAM) | 2 µM | 2 µM | 1.6 µM | 2 µM |
| RV(5′-ph) | 2 µM | - | - | - |
| RV(pA) | - | 2 µM | - | 0.04 µM |
| RV | - | - | 0.08 µM | - |
| RV(bl) | - | - | - | 1.96 µM |
| Stage | PCR-Lambda PCR-Long RV A-PCR | PBA-PCR | |
|---|---|---|---|
| First denaturation | 95°C—5′ | 95°C—5′ | |
| Denaturation | PCR | 95°C—10″ | 95°C—10″ |
| Annealing | 60°C—10″ | 60°C—30″ | |
| Elongation | 72°C—10″ | 72°C—20″ | |
| Final elongation | 72°C—30″ | 72°C—30″ | |
| Cycles | PCR-lambda | PCR-long RV | A-PCR | PBA-PCR |
|---|---|---|---|---|
| 10 | 100 | 100 | 48.6 | 100 |
| 15 | 68.7 | 40.1 | 53.0 | 89.7 |
| 20 | 49.8 | 22.0 | 69.3 | 90.5 |
| 25 | 38.1 | 13.4 | 71.7 | 90.5 |
| Average | 51.3 | 35.1 | 48.5 | 92.7 |
| PCR-lambda | PCR-long RV | A-PCR | PBA-PCR | |
|---|---|---|---|---|
| Specificity (relative amount of specific product, %) | 51.3 | 35.1 | 48.5 | 92.7 |
| Efficacy (yield of purified ssDNA ± RMSD, ng) | 110.5 ± 33.6 | 88.2 ± 30.6 | 191.4 ± 16.8 | 553.6 ± 93.7 |
| Variation Coefficient, V | 0.31 | 0.35 | 0.09 | 0.17 |
| Time consumed (protocol duration, hours) | 11.5 | 9.5 | 8.5 | 9.0 |
| Specific reagents | Lambda Exonuclease; RV(5′-ph) | RV(dA); dPAGE | - | RV(dA); RV(bl) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garanin, A.; Shalaev, A.; Zabegina, L.; Kadantseva, E.; Sharonova, T.; Malek, A. Specific Aspects of SELEX Protocol: Different Approaches for ssDNA Generation. Methods Protoc. 2025, 8, 36. https://doi.org/10.3390/mps8020036
Garanin A, Shalaev A, Zabegina L, Kadantseva E, Sharonova T, Malek A. Specific Aspects of SELEX Protocol: Different Approaches for ssDNA Generation. Methods and Protocols. 2025; 8(2):36. https://doi.org/10.3390/mps8020036
Chicago/Turabian StyleGaranin, Alexandr, Andrey Shalaev, Lidia Zabegina, Ekaterina Kadantseva, Tatiana Sharonova, and Anastasia Malek. 2025. "Specific Aspects of SELEX Protocol: Different Approaches for ssDNA Generation" Methods and Protocols 8, no. 2: 36. https://doi.org/10.3390/mps8020036
APA StyleGaranin, A., Shalaev, A., Zabegina, L., Kadantseva, E., Sharonova, T., & Malek, A. (2025). Specific Aspects of SELEX Protocol: Different Approaches for ssDNA Generation. Methods and Protocols, 8(2), 36. https://doi.org/10.3390/mps8020036