A Streamlined Approach to Pathway Analysis from RNA-Sequencing Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
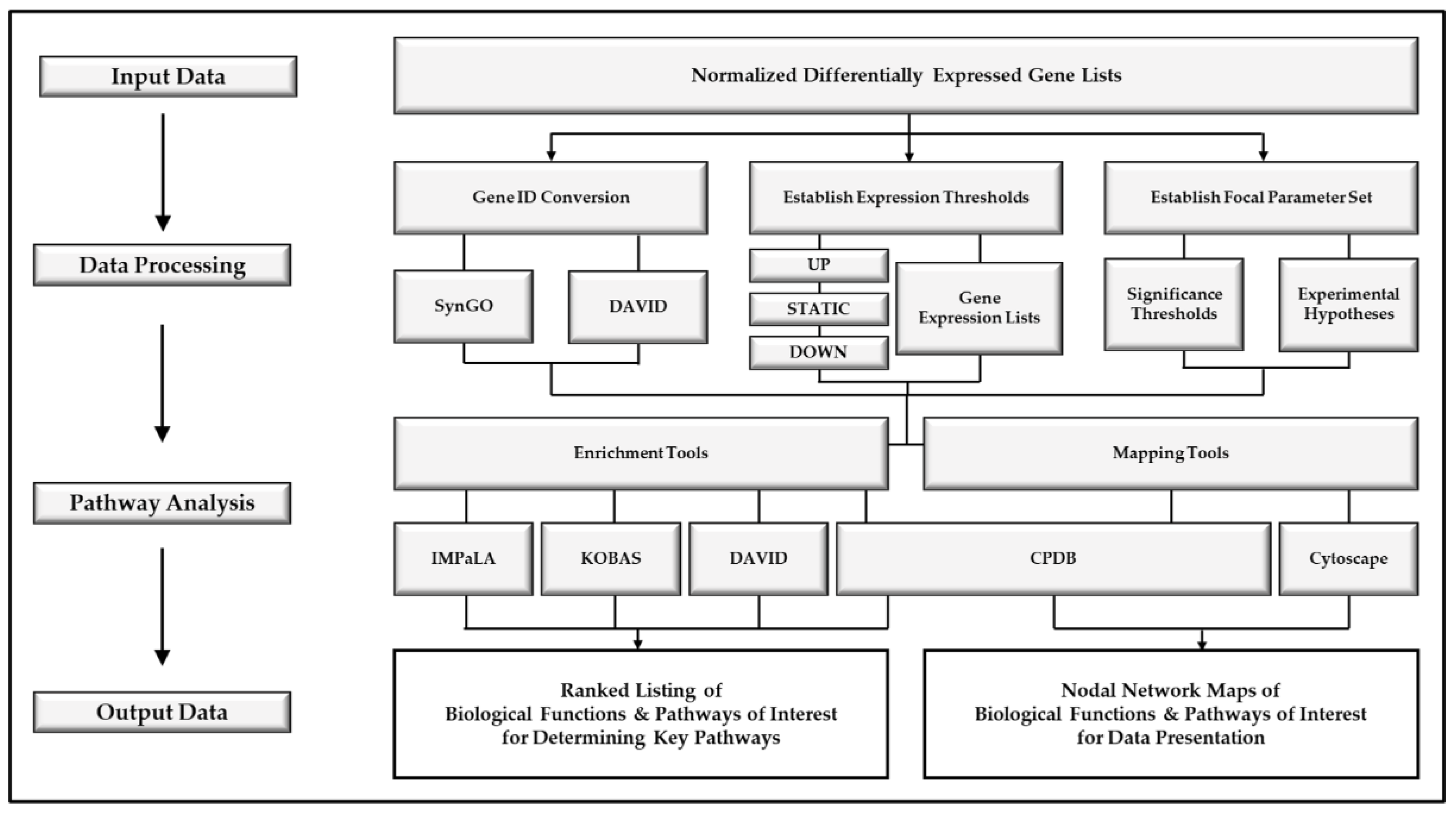
2. Experimental Design
2.1. Data Processing
2.2. Pathway Analysis Tools
2.3. Nodal Network Generation
3. Procedure
3.1. Data Processing
- Locate and open the files containing the differentially expressed gene lists for a sample comparison.
- Copy and paste provided gene identifiers directly into SynGO conversion tool [7] and select corresponding identifier type and species from drop-down menus.
- Press Start ID Conversion and download resulting table to text file.
- Import text file to new spreadsheet and integrate data with differentially expressed gene list document resulting in enhanced gene identifier options for downstream enrichment programs.
- Establish expression thresholds for gene sets to clearly define upregulated, downregulated, and static expression sets (this can be based on pre-existing thresholds provided by company or modified based on relevant literature).
- Segment master gene list to three separate spreadsheets based the classification of whether the gene is upregulated, downregulated, or statically expressed between the experimental group and respective control in comparison.
- Repeat steps 1–6 for all comparisons to be examined.
- For each comparison set, determine biological functions/pathways most likely to be impacted based on the overarching hypothesis for the study (i.e., samples derived from cells treated with a compound intending to enhance osteogenesis would be examined focally for variations in osteoblastic differentiation or cellular adhesion pathways) and classify these functions/pathways as the focal parameter set.
- Record the established focal parameter set for use in later enrichment steps.
- Open a blank spreadsheet and designate as “Enrichment Analysis Master List” (this will be the repository for downstream enrichment data results from pathway analysis tools).
3.2. Pathway Analysis
3.2.1. IMPaLA Analysis
- Open IMPaLA homepage [50].
- Input gene identifiers and respective expression values, select gene identifier type from drop-down menu, select Wilcoxon pathway enrichment analysis, and press Start Analysis.
- Verify that an adequate percentage of the genes were detected and mapped (a low percentage may indicate input error).
- Sort resulting table by Qgenes and download pathway list as a spreadsheet; see Figure 1.
- Remove list items above designated significance threshold based on Qgenes value and transfer remaining pathways to “Enrichment Analysis Master List”.
3.2.2. KOBAS Analysis
- Open KOBAS homepage [51] and navigate to gene enrichment analysis (Enrichment → Gene-list Enrichment).
- Enter target species, select gene identifier type for menu (note that KOBAS is capable of accepting Fasta type files), input gene identifiers, select KEGG Pathway (K), Reactome (R), GO (G) database options, and press Run.
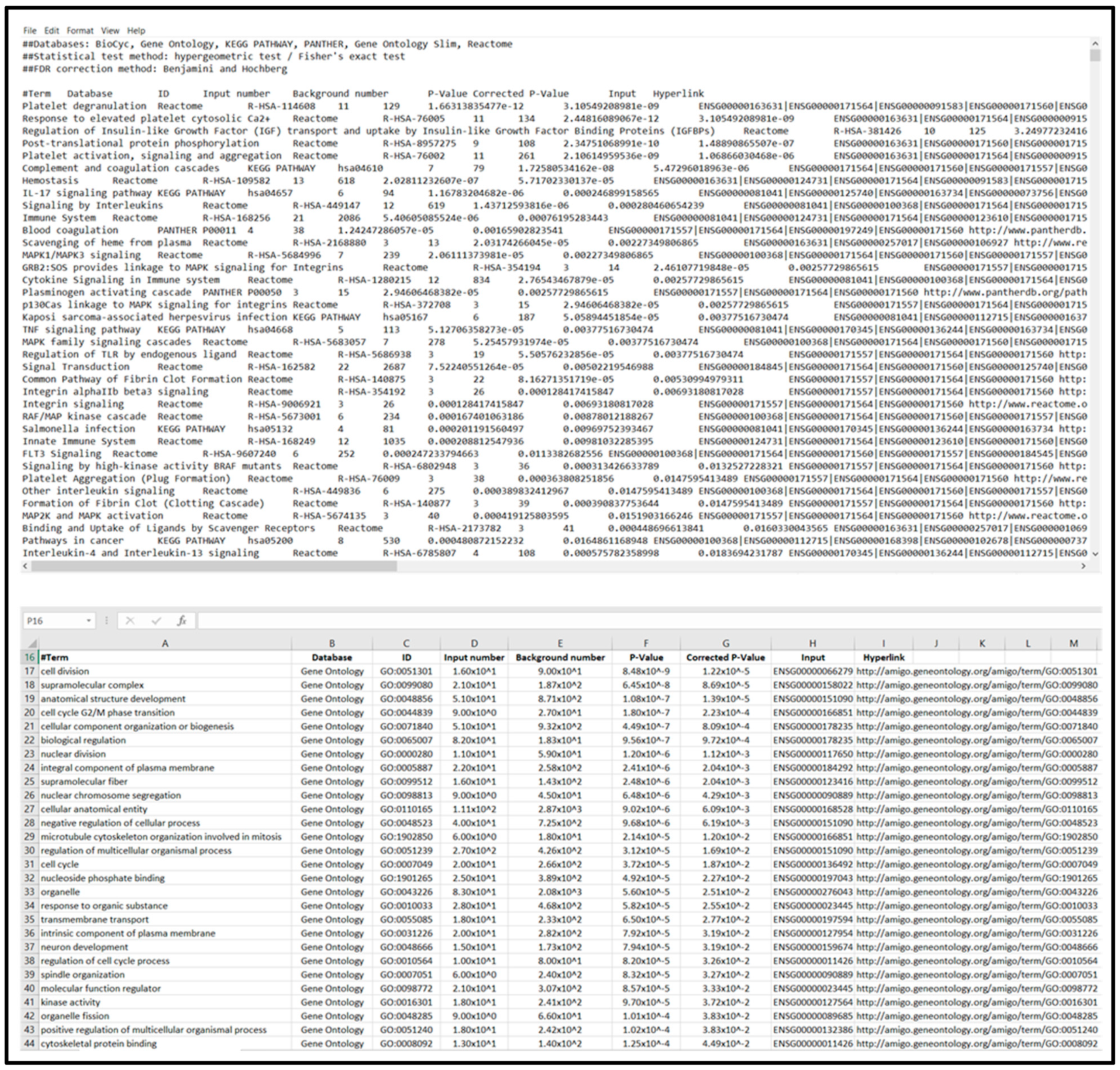
- Sort table items by corrected p-value and download list using Download Total Terms function to generate a text file that can be imported to “Enrichment Analysis Master List”; see Figure 2.
- Click Visualization of Filtered Terms (note that this function is currently in demo state and will likely be improved in the future) to generate circular network, barplot, and bubble plot representations of data.
- Save graphics of interest with the save feature at bottom right of each image (these graphics can be stored for supplemental data).
3.2.3. DAVID Analysis
- Input gene identifiers, select identifier type from drop-down menu, specify list as gene list, and press Submit List.
- Select species to filter output data (In Gene List Manager tab select the target species name → click Select Species → for Select List to: click Use)
- Select Functional Annotation Tool and utilize DAVID default search criteria (Search criteria can be modified to further expand pathways/biological functions examined. To edit the search parameters, such as utilized pathway databases, expand annotation categories and select/deselect search parameters).
- Open Functional Annotation Clustering and download cluster data to text file for importing to “Enrichment Analysis Master List”; see Figure 3.
- For saving heatmaps of high enrichment score clusters, open heatmap of interest and print resulting webpage to PDF for supplemental data file.
3.2.4. CPDB Analysis
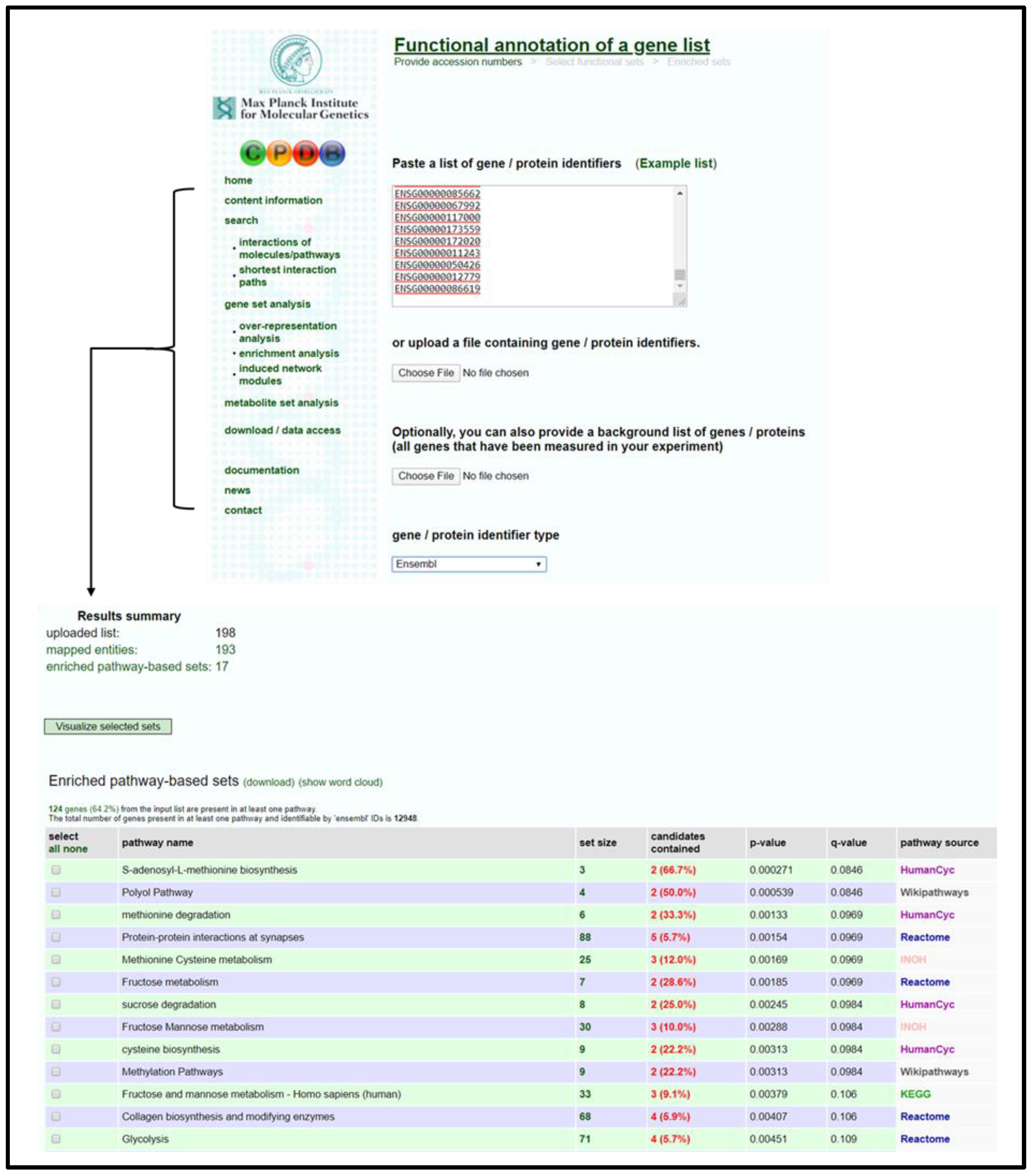
- Open CPDB homepage [53] and navigate to gene enrichment analysis (Gene Set Analysis → Enrichment Analysis).
- Input gene identifiers and respective expression values, select identifier type from drop-down menu, and press proceed.
- Verify that an adequate percentage of the genes were detected and mapped (a low percentage may indicate input error).
- Select 1-next neighbor, pathways as defined by pathway databases, gene ontology level 2 categories, and sets of genes whose products are found together in protein complexes from the following page to establish the extent of the enrichment analysis.
- Sort the resulting list by q-value and select pathways/biological functions relating to focal parameter set; see Figure 4.
- Download selected pathway/biological functions to text file and import to “Enrichment Analysis Master List”.
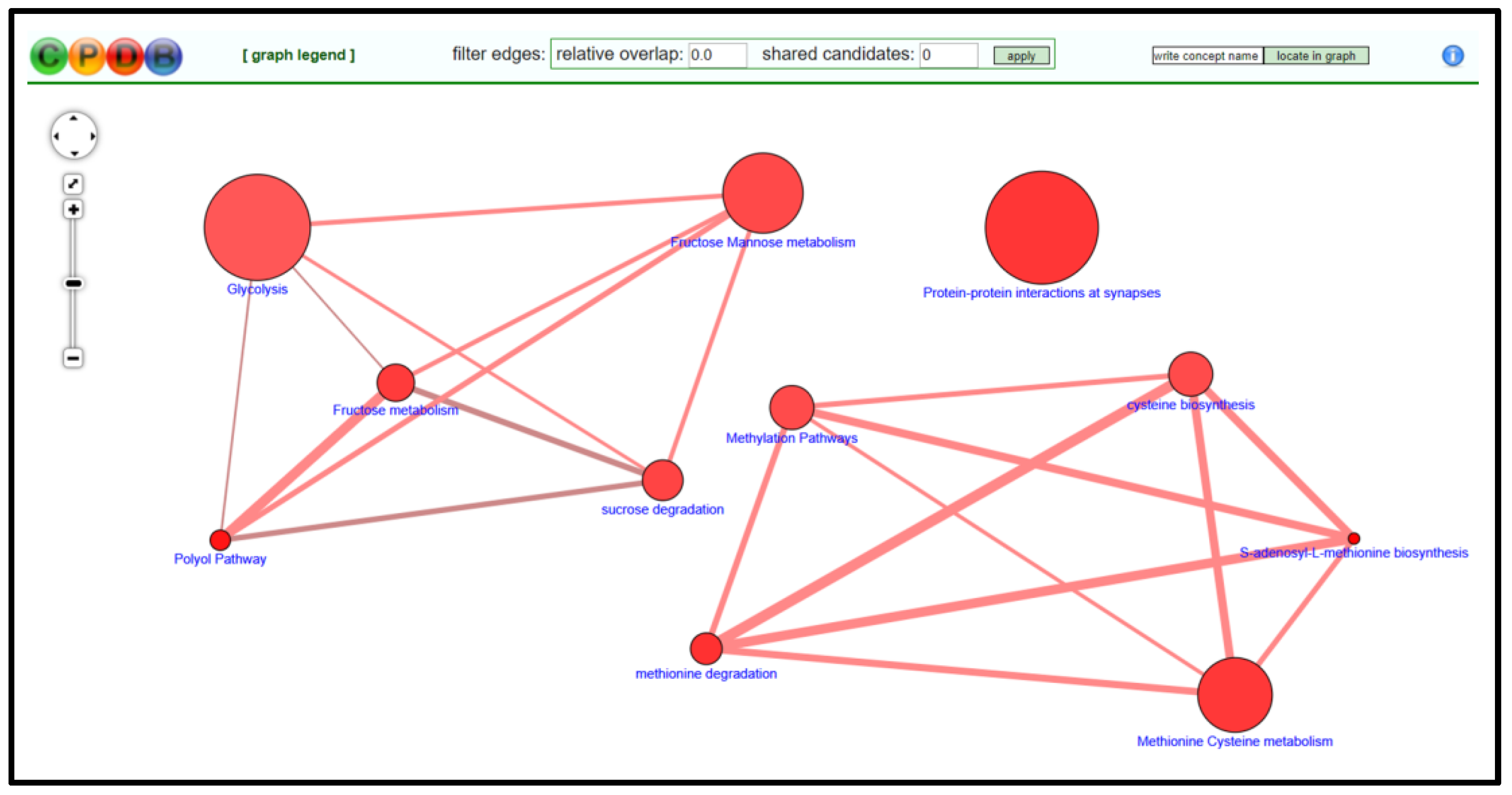
- On CPDB, press visualize selected sets to generate a nodal network detailing the connective elements among selected pathways/biological functions; see Figure 5.
- Modify node position and connective edge filter settings to best display data.
- Select graph legend to insert legend onto network image.
- Capture network image with screen print function and edit/save final graphic using PowerPoint software.
3.2.5. Enrichment Analysis Cross-Referencing
- Organize data within “Enrichment Analysis Master List” spreadsheet so that pathways/biological functions and respective corrected significance values from IMPaLA, KOBAS, DAVID, and CPDB are in side-by-side lists.
- Sort each list by corrected significance value and remove all line items above designated significance threshold.
- Based on focal parameter set, highlight all PoIs using the cell highlighting tool in spreadsheet application.
- Sort each list by cell color (this will bring all highlighted cells to the top of lists) and remove unhighlighted line items.
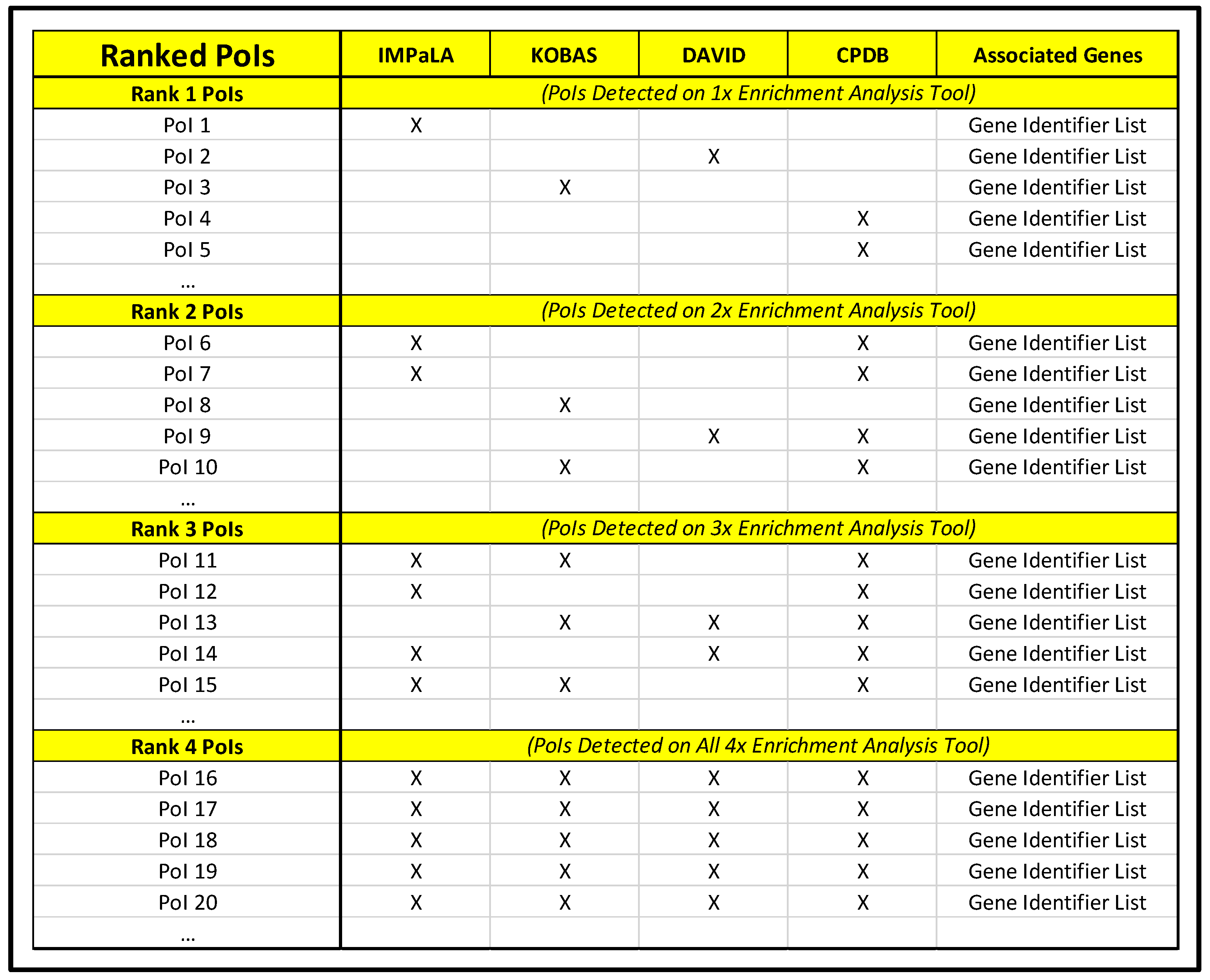
- Manually compare lists for commonalities and highlight with a new color any PoIs shared by two or more pathway analysis tools (using a different color to denote that a PoI is common to two, three, or four pathway analysis tools).
- Generate a new document designated as “Ranked PoIs” categorizing PoIs by commonality among pathway analysis tools and sorted by corrected significance values.
- Repeat steps 3.2.1–3.2.5 for the upregulated, downregulated, and statically expressed gene lists for each comparison being assessed.
- For each set of upregulated, downregulated, and statically expressed gene lists, construct composition spreadsheet for top-ranked PoIs (those observed across multiple pathway analysis tools) that includes a column of gene symbol names for all genes associated with selected PoIs and columns headed with each PoI name for developing a search index in Cytoscape (entering YES in the PoI name column for genes associated with it will permit rapid categorization downstream).


3.2.6. Cytoscape Mapping
- Open Cytoscape homepage [54] and download the most recent version of Cytoscape software (can periodically check homepage for updated versions if already downloaded).
- Install stringApp application through in-software or online app manager (other plug-ins can be install and explored for effectiveness in modifying data).
- Open BioGRID homepage [55] and download the most recent zip file for “BIOGRID-ORGANISM-(Update Number).psi25”.
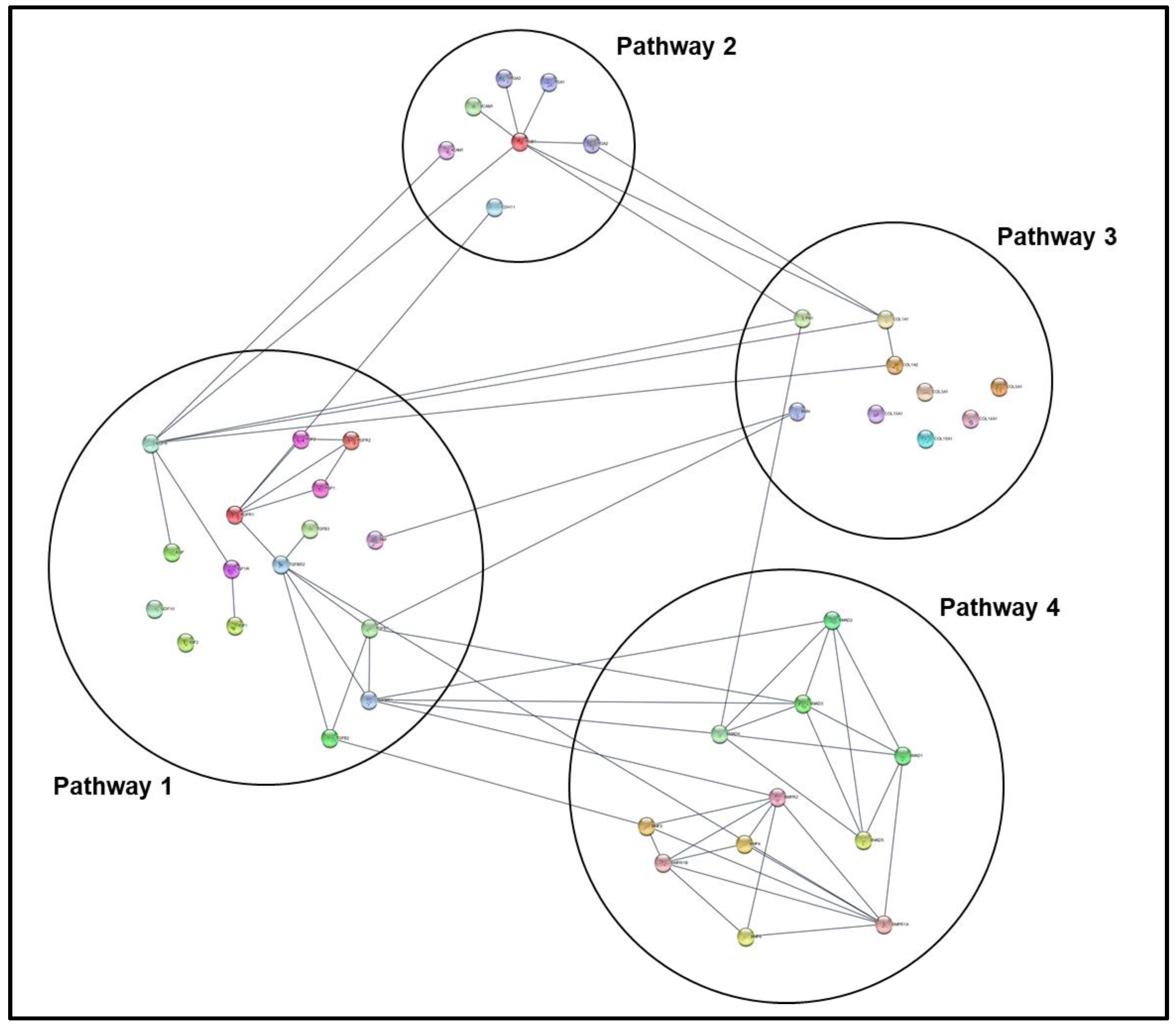
- Import BioGRID Homo Sapiens XML document from zip file into Cytoscape (File → Import → Network from File → Select Homo Sapiens XML Document); see Figure 6.
- Import upregulated, downregulated, and statically expressed gene lists for target comparison and overlay with BioGRID basal network (File → Import → Table from File → Select Gene List Name → Select To selected networks only for Where to Import Table Data → Select BioGRID basal network from Network List → Select Node Table Columns for Import Data as → Select Short Label for Key Column for Networks → Keep Case Sensitive Key Values selected → Find the gene symbol name column of gene list and click on column to edit → Set as Key with the key shaped icon under Meaning in the drop-down menu → Press OK to import and overlay data).
- Import composition spreadsheet for data set and overlay with network for PoI search index (File → Import → Table from File → Select Composition Spreadsheet Name → Select To selected networks only for Where to Import Table Data → Select BioGRID basal network from Network List → Select Node Table Columns for Import Data as → Select Short Label for Key Column for Networks → Keep Case Sensitive Key Values selected → Find the gene symbol name column of composition spreadsheet and click on column to edit → Set as Key with the key shaped icon under Meaning in the drop-down menu → Press OK to import and overlay data).
- Highlight gene list data nodes from newly constructed network by selecting them from Node Table (sorting the table by a column of values not in the original BioGRID data such as the entrez ID or HGNC ID will bring all gene list data to the top of table).
- Generate new network from selected nodes and edges for data-specific nodal network (File → New Network → From Selected Nodes, Selected Edges).
- Remove duplicate edges (Edit → Remove Duplicate Edges… → Select target network and press OK).
- Remove self-loops (Edit → Remove Self-loops… → Select target network and press OK).
- Clone data-specific network to maintain unmodified version of network for reference (File → New Network → Clone Current Network).
- Add color gradient to editable data-specific network nodes based on associated expression value to establish (Style tab → Fill Color drop-down menu → Select expression fold change value column for Column → Select Continuous Mapping for Mapping Type → Select colors for low, high, and static expression to form gradient → Press OK to apply).
- Separate color-coded network nodes by associated PoI using search index columns in node table (sort each PoI column to isolate nodes associated with it, then click and drag these clusters to independent regions on the mapping panel).
- Organize PoI clusters either manually or using one of the automated formatting options under the Layout tab.
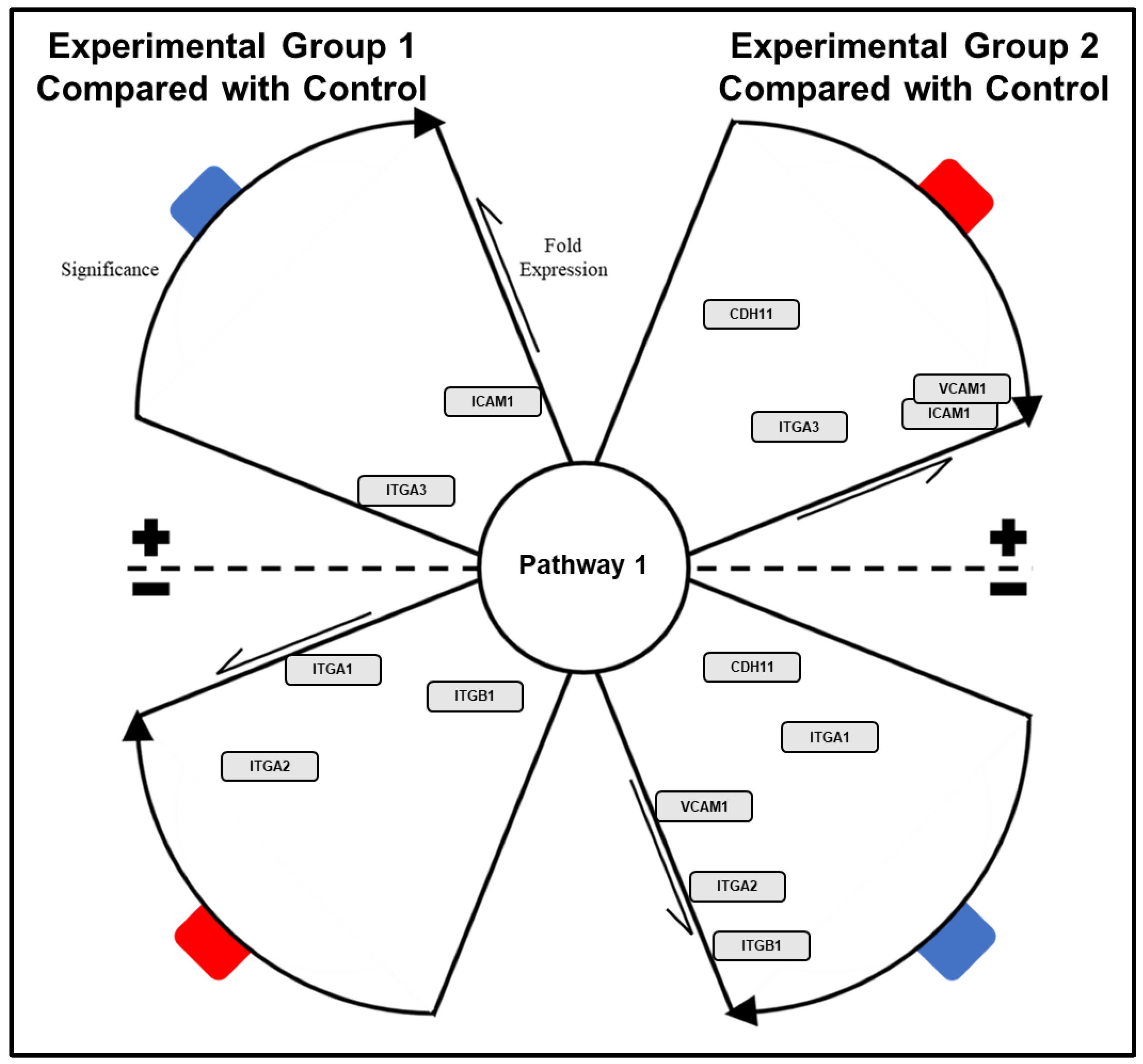
- Annotate resulting node map with figures and text as needed using the Annotation tab.
- For protein-protein annotation data and enhanced node graphics, clone the current network (covered in a previous step) and apply the stringApp plug-in to the resulting map (this will replace nodes with high resolution graphics of the associated protein structure and add protein-protein data to network annotation, but may reorganize network orientation).
- Once annotation and organization of nodal network is complete, export network map as image for use in presentations/publications (File → Export → Network to Image → Save as .PNG file for high resolution).
4. Expected Results
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kornobis, E.; Cabellos, L.; Aguilar, F.; Frías-López, C.; Rozas, J.; Marco, J.; Zardoya, R. TRUFA: A User-Friendly Web Server for de novo RNA-seq Analysis Using Cluster Computing. Evol. Bioinform. 2015, 11, EBO.S23873. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Torson, A.S.; Dong, Y.-W.; Sinclair, B.J. Help, there are ‘omics’ in my comparative physiology! J. Exp. Biol. 2020, 223, 191262. [Google Scholar]
- Chen, L.; Fei, C.; Zhu, L.; Xu, Z.; Zou, W.; Yang, T.; Lin, H.; Xi, D. RNA-seq approach to analysis of gene expression profiles in dark green islands and light green tissues of Cucumber mosaic virus-infected Nicotiana tabacum. PLoS ONE 2017, 12, e0175391. [Google Scholar] [CrossRef]
- Warden, C.D.; Yuan, Y.-C.; Wu, X. Optimal calculation of RNA-Seq fold-change values. Int. J. Comput. Bioinform. Silico Model. 2013, 2, 285–292. [Google Scholar]
- DAVID Gene ID Conversion Tool. Available online: https://david.ncifcrf.gov/conversion.jsp (accessed on 15 March 2021).
- SYNGO ID Conversion Tool. Available online: https://syngoportal.org/convert.html (accessed on 15 March 2021).
- Koopmans, F.; van Nierop, P.; Andres-Alonso, M.; Byrnes, A.; Cijsouw, T.; Coba, M.P.; Cornelisse, L.N.; Farrell, R.J.; Goldschmidt, H.L.; Howrigan, D.P.; et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 2019, 103, 217–234.e4. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Gene Annotation and Pathway Mapping in KEGG. Methods Mol. Biol. 2007, 396, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef]
- The Gene Ontology Resource: 20 years and still Going strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.-S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.D.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Li, C.; Han, J.; Yao, Q.; Zou, C.; Xu, Y.; Zhang, C.; Shang, D.; Zhou, L.; Zou, C.; Sun, Z.; et al. Subpathway-GM: Identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic Acids Res. 2013, 41, e101. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank-Quinn, C.I.; Jacobson, S.; Hughes, G.; Powell, R.L.; Petrache, I.; Kechris, K.; Bowler, R.; Reisdorph, N. Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci. Rep. 2018, 8, 17132. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, S.; Li, L.; Zhang, L.; Xian, S.; Wang, Y.; Cheng, Y. Identification of differentially expressed genes and signaling pathways in ovarian cancer by integrated bioinformatics analysis. OncoTargets Ther. 2018, 11, 1457–1474. [Google Scholar] [CrossRef]
- Shen, S.; Kong, J.; Qiu, Y.; Yang, X.; Wang, W.; Yan, L. Identification of core genes and outcomes in hepatocellular carcinoma by bioinformatics analysis. J. Cell. Biochem. 2019, 120, 10069–10081. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, L.; Shi, L.; Yun, F.; Liu, X.; Chen, Y.; Chen, C.; Ren, Y.; Jia, Y. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 2019, 24, 38. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Feng, H.; Gu, Z.-Y.; Li, Q.; Liu, Q.-H.; Yang, X.-Y.; Zhang, J.-J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yuan, Q.; Wu, D.; Meng, X.; Wang, B. Identification of core genes and outcome in gastric cancer using bioinformatics analysis. Oncotarget 2017, 8, 70271–70280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Witten, D.M.; Johnstone, I.M.; Tibshirani, R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics 2011, 13, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.K.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2010, 39, D712–D717. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012, 41, D793–D800. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Demchak, B.; Otasek, D.; Pico, A.R.; Bader, G.D.; Ono, K.; Settle, B.; Sage, E.; Morris, J.H.; Longabaugh, W.; Lopes, C.; et al. The Cytoscape Automation app article collection. F1000Research 2018, 7, 800. [Google Scholar] [CrossRef]
- Huang, L.J.; Law, J.N.; Murali, T.M. Automating the PathLinker app for Cytoscape. F1000Research 2018, 7, 727. [Google Scholar] [CrossRef]
- Mustafin, Z.S.; Lashin, S.A.; Matushkin, Y.G.; Gunbin, K.V.; Afonnikov, D.A. Orthoscape: A cytoscape application for grouping and visualization KEGG based gene networks by taxonomy and homology principles. BMC Bioinform. 2017, 18 (Suppl. 1), 1427. [Google Scholar] [CrossRef][Green Version]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2013, 13, 2498–2504. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Treister, A.; Pico, A.R. Identifier Mapping in Cytoscape. F1000Research 2018, 7, 725. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, F.; Luo, S.; Yin, X.; He, D.; Liu, J.; Yue, Z.; Song, J. Co-expression of key gene modules and pathways of human breast cancer cell lines. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef]
- Politano, G.; Benso, A.; Savino, A.; Di Carlo, S. ReNE: A Cytoscape Plugin for Regulatory Network Enhancement. PLoS ONE 2014, 9, e115585. [Google Scholar] [CrossRef]
- Wolfien, M.; Rimmbach, C.; Schmitz, U.; Jung, J.J.; Krebs, S.; Steinhoff, G.; David, R.; Wolkenhauer, O. TRAPLINE: A stand-ardized and automated pipeline for RNA sequencing data analysis, evaluation and annotation. BMC Bioinform. 2016, 17, 21. [Google Scholar] [CrossRef]
- Bebek, G. Identifying gene interaction networks. Breast Cancer 2011, 850, 483–494. [Google Scholar]
- Merico, D.; Gfeller, D.; Bader, G.D. How to visually interpret biological data using networks. Nat. Biotechnol. 2009, 27, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Vijay, D.; Federowicz, S.; Pico, A.R.; Ferrin, T.E. CyAnimator: Simple Animations of Cytoscape Networks. F1000Research 2015, 4, 482. [Google Scholar] [CrossRef] [PubMed]
- Integrated Molecular Pathway Level Analysis. Available online: http://impala.molgen.mpg.de/ (accessed on 15 March 2021).
- KEGG Orthology-Based Annotation System. Available online: http://kobas.cbi.pku.edu.cn/kobas3 (accessed on 15 March 2021).
- DAVID Bioinformatics Resources 6.8. Available online: https://david.ncifcrf.gov/home.jsp (accessed on 15 March 2021).
- ConsensusPathDB. Available online: http://cpdb.molgen.mpg.de/ (accessed on 15 March 2021).
- Cytoscape. Available online: https://cytoscape.org/ (accessed on 15 March 2021).
- BioGRID. Available online: https://thebiogrid.org/ (accessed on 15 March 2021).








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bow, A. A Streamlined Approach to Pathway Analysis from RNA-Sequencing Data. Methods Protoc. 2021, 4, 21. https://doi.org/10.3390/mps4010021
Bow A. A Streamlined Approach to Pathway Analysis from RNA-Sequencing Data. Methods and Protocols. 2021; 4(1):21. https://doi.org/10.3390/mps4010021
Chicago/Turabian StyleBow, Austin. 2021. "A Streamlined Approach to Pathway Analysis from RNA-Sequencing Data" Methods and Protocols 4, no. 1: 21. https://doi.org/10.3390/mps4010021
APA StyleBow, A. (2021). A Streamlined Approach to Pathway Analysis from RNA-Sequencing Data. Methods and Protocols, 4(1), 21. https://doi.org/10.3390/mps4010021