Unexpectedly High Levels of Inverted Re-Insertions Using Paired sgRNAs for Genomic Deletions

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
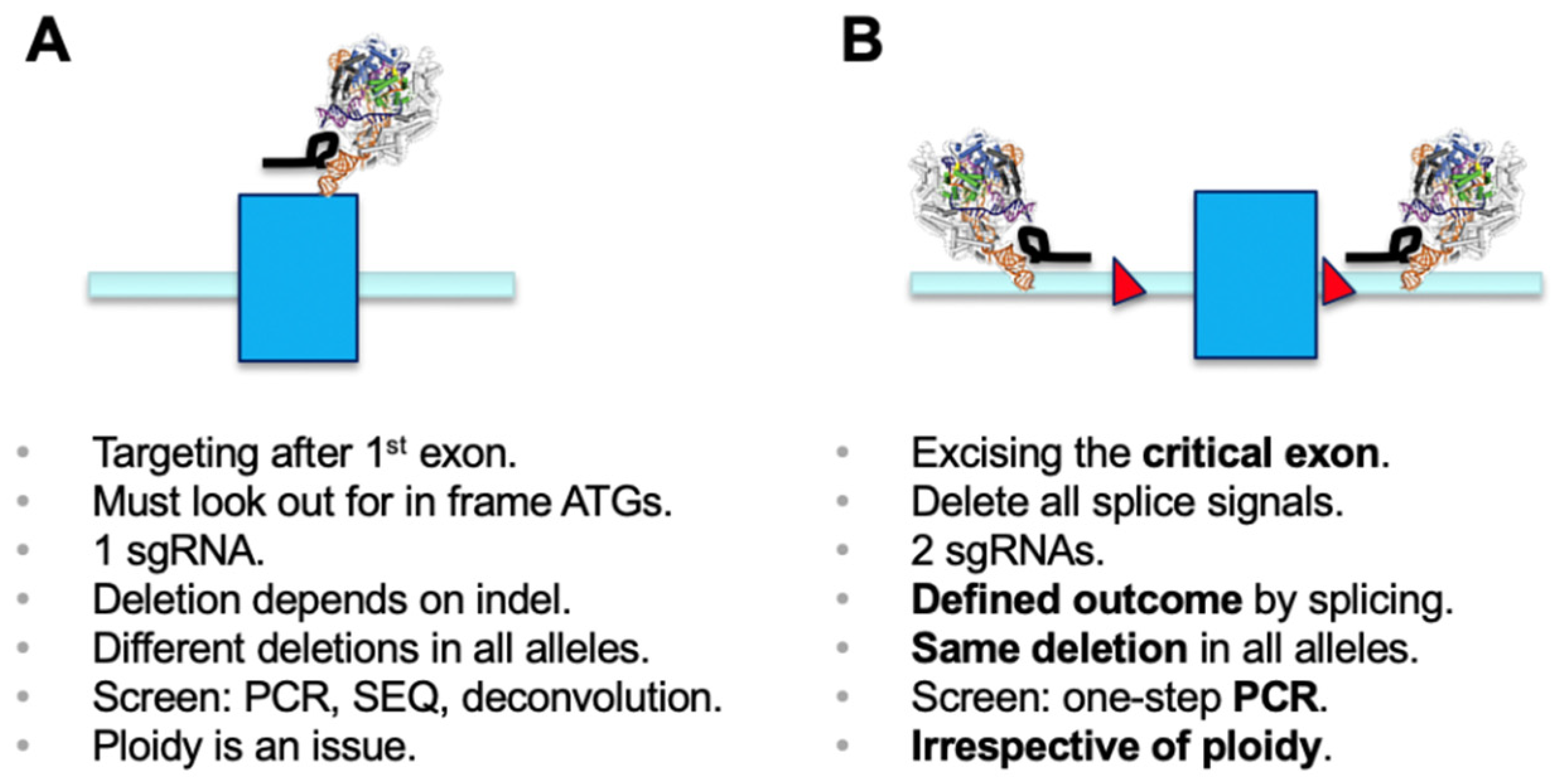
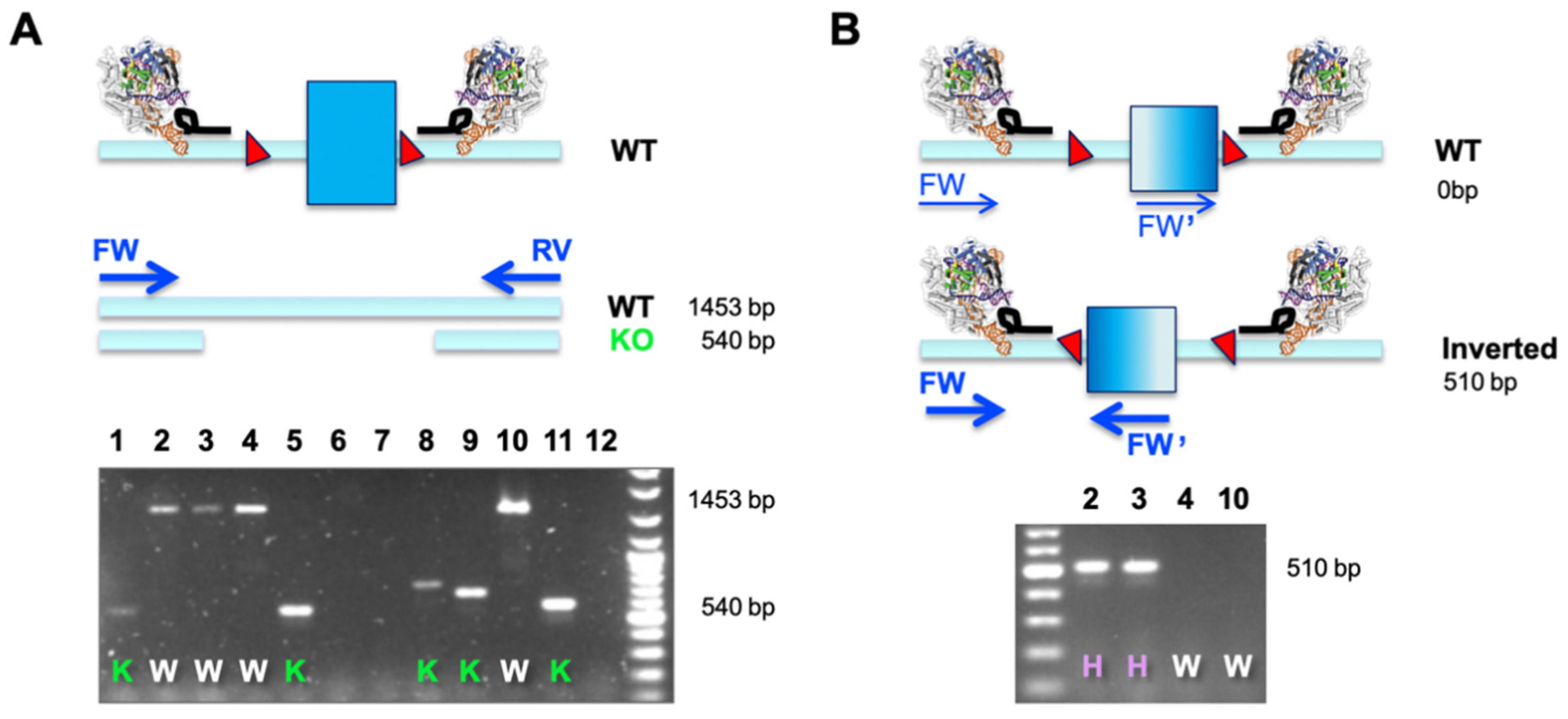
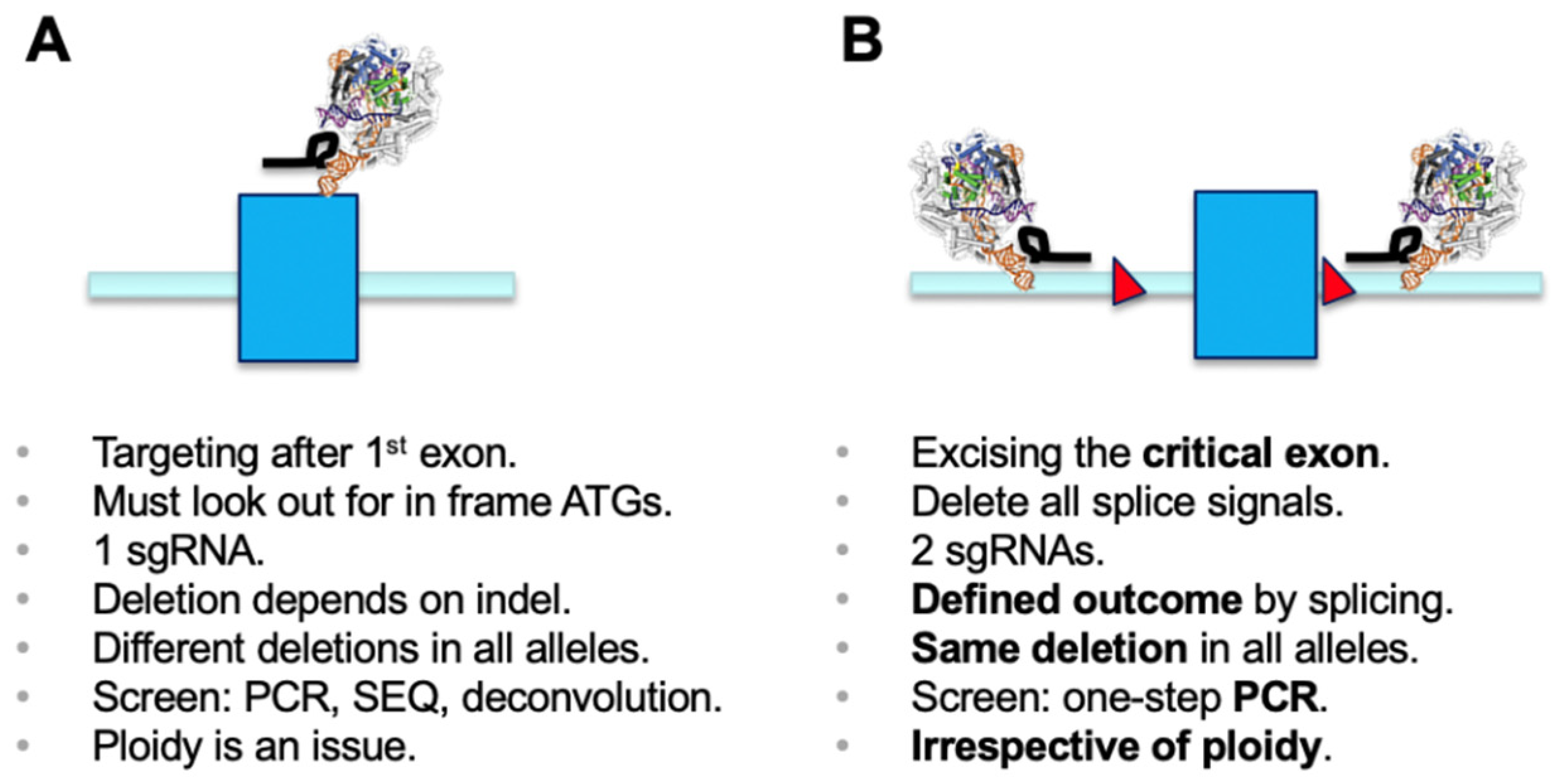
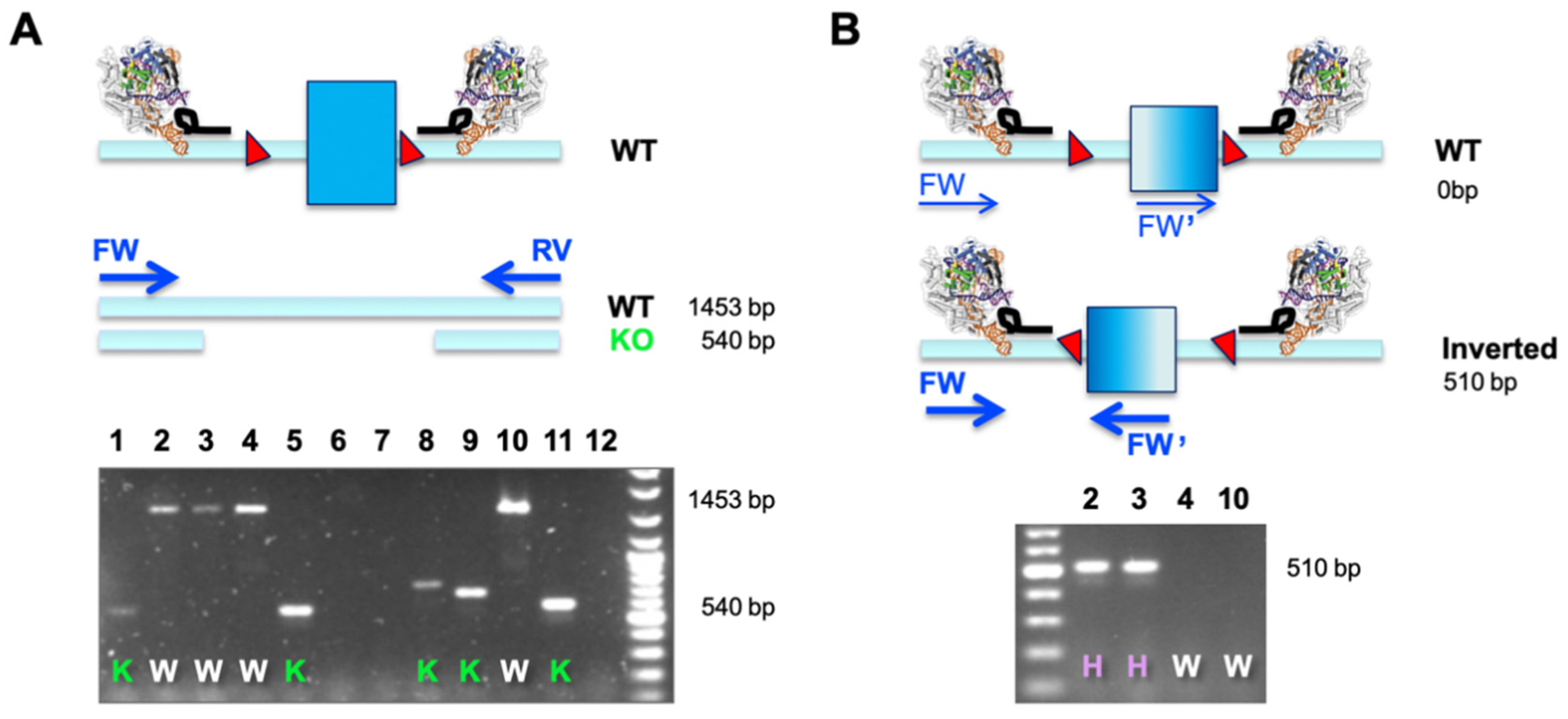
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Lonowski, L.A.; Narimatsu, Y.; Riaz, A.; Delay, C.E.; Yang, Z.; Niola, F.; Duda, K.; Ober, E.A.; Clausen, H.; Wandall, H.H.; et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protoc. 2017, 12, 581–603. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic. Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, J.; Shen, B.; Chen, L.; Su, Y.; Yang, J.; Zhang, W.; Tian, X.; Huang, X. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 2014, 281, 1717–1725. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, J.; Xiong, M.; Petersen, A.J.; Dong, Y.; Tao, Y.; Huang, C.T.; Du, Z.; Zhang, S.C. Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell 2015, 17, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, C.; Liu, W.; Gao, W.; Liu, C.; Song, G.; Li, W.X.; Mao, L.; Chen, B.; Xu, Y.; et al. An alternative strategy for targeted gene replacement in plants using a dual-sgRNA/Cas9 design. Sci. Rep. 2016, 6, 23890. [Google Scholar] [CrossRef]
- Mettananda, S.; Fisher, C.A.; Hay, D.; Badat, M.; Quek, L.; Clark, K.; Hublitz, P.; Downes, D.; Kerry, J.; Gosden, M.; et al. Editing an alpha-globin enhancer in primary human hematopoietic stem cells as a treatment for beta-thalassemia. Nat. Commun. 2017, 8, 424. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencia, A.; Garcia, M.; Torres, R.; Rodriguez, S.; Carretero, M.; Chacon-Solano, E.; Modamio-Hoybjor, S.; Marinas, L.; Leon, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, K.; Doe, B.; Banerjee, R.; Yang, F.; Bradley, A. Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 2016, 54, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birling, M.C.; Schaeffer, L.; Andre, P.; Lindner, L.; Marechal, D.; Ayadi, A.; Sorg, T.; Pavlovic, G.; Herault, Y. Efficient and rapid generation of large genomic variants in rats and mice using CRISMERE. Sci. Rep. 2017, 7, 43331. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.Y.; Wang, C.; Lee, H.K.; Yoo, K.H.; Zeng, X.; Kuhns, T.; Yang, C.M.; Mohr, T.; Liu, C.; Hennighausen, L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017, 8, 15464. [Google Scholar] [CrossRef]
- Boroviak, K.; Fu, B.; Yang, F.; Doe, B.; Bradley, A. Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci. Rep. 2017, 7, 12867. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Owens, D.D.G.; Caulder, A.; Frontera, V.; Harman, J.R.; Allan, A.J.; Bucakci, A.; Greder, L.; Codner, G.F.; Hublitz, P.; McHugh, P.J.; et al. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic. Acids Res. 2019, 47, 7402–7417. [Google Scholar] [CrossRef] [Green Version]
- Skryabin, B.V.; Kummerfeld, D.M.; Gubar, L.; Seeger, B.; Kaiser, H.; Stegemann, A.; Roth, J.; Meuth, S.G.; Pavenstadt, H.; Sherwood, J.; et al. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9-mediated genome editing events. Sci. Adv. 2020, 6, eaax2941. [Google Scholar] [CrossRef] [Green Version]
- Korablev, A.; Lukyanchikova, V.; Serova, I.; Battulin, N. On-Target CRISPR/Cas9 Activity Can Cause Undesigned Large Deletion in Mouse Zygotes. Int. J. Mol. Sci. 2020, 21, 3604. [Google Scholar] [CrossRef]
- Przewrocka, J.; Rowan, A.; Rosenthal, R.; Kanu, N.; Swanton, C. Unintended on-target chromosomal instability following CRISPR/Cas9 single gene targeting. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wrona, D.; Pastukhov, O.; Pritchard, R.S.; Raimondi, F.; Tchinda, J.; Jinek, M.; Siler, U.; Reichenbach, J. CRISPR-Directed Therapeutic Correction at the NCF1 Locus Is Challenged by Frequent Incidence of Chromosomal Deletions. Mol. Ther. Methods Clin. Dev. 2020, 17, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, I.; Kroeger, J.A.; Malik, R.; Klimmt, J.; Crusius, D.; Dannert, A.; Dichgans, M.; Paquet, D. Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep. 2020, 31, 107689. [Google Scholar] [CrossRef] [PubMed]
- Teboul, L.; Herault, Y.; Wells, S.; Qasim, W.; Pavlovic, G. Variability in Genome Editing Outcomes: Challenges for Research Reproducibility and Clinical Safety. Mol. Ther. 2020, 28, 1422–1431. [Google Scholar] [CrossRef]
- Haeussler, M.; Schonig, K.; Eckert, H.; Eschstruth, A.; Mianne, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef]
- Cocks, G.; Curran, S.; Gami, P.; Uwanogho, D.; Jeffries, A.R.; Kathuria, A.; Lucchesi, W.; Wood, V.; Dixon, R.; Ogilvie, C.; et al. The utility of patient specific induced pluripotent stem cells for the modelling of Autistic Spectrum Disorders. Psychopharmacology (Berl.) 2014, 231, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Hooper, M.; Hardy, K.; Handyside, A.; Hunter, S.; Monk, M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 1987, 326, 292–295. [Google Scholar] [CrossRef]
- Yu, X.; Liang, X.; Xie, H.; Kumar, S.; Ravinder, N.; Potter, J.; de Mollerat du Jeu, X.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Mianne, J.; Bourguignon, C.; Nguyen Van, C.; Fieldes, M.; Nasri, A.; Assou, S.; De Vos, J. Pipeline for the Generation and Characterization of Transgenic Human Pluripotent Stem Cells Using the CRISPR/Cas9 Technology. Cells 2020, 9, 1312. [Google Scholar] [CrossRef]
- Kwon, M.J.; Ju, T.J.; Heo, J.Y.; Kim, Y.W.; Kim, J.Y.; Won, K.C.; Kim, J.R.; Bae, Y.K.; Park, I.S.; Min, B.H.; et al. Deficiency of clusterin exacerbates high-fat diet-induced insulin resistance in male mice. Endocrinology 2014, 155, 2089–2101. [Google Scholar] [CrossRef] [Green Version]
- De Vree, P.J.; de Wit, E.; Yilmaz, M.; van de Heijning, M.; Klous, P.; Verstegen, M.J.; Wan, Y.; Teunissen, H.; Krijger, P.H.; Geeven, G.; et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat. Biotechnol. 2014, 32, 1019–1025. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, V.; Shen, B.; Zhang, W.; Hodgkins, A.; Keane, T.; Huang, X.; Skarnes, W.C. Off-target mutations are rare in Cas9-modified mice. Nat. Methods 2015, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Boroviak, K.; Thomas, M.; Doe, B.; Riva, L.; Ryder, E.; Adams, D.J. No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 2018, 14, e1007503. [Google Scholar] [CrossRef]
- Willi, M.; Smith, H.E.; Wang, C.; Liu, C.; Hennighausen, L. Mutation frequency is not increased in CRISPR-Cas9-edited mice. Nat. Methods 2018, 15, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Tsui, C.K.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017, 8, 15178. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cell line | Target | Total | WT/HET | Δ | %Δ | INV | %INV | |
|---|---|---|---|---|---|---|---|---|
| 293 | Exon PEX5 | 5 | 4 | 1 | 20 | 1 | 20 | |
| 293 T-Rex | Exon PEX5 | 26 | 18 | 8 | 31 | 5 | 19 | |
| 293 | Exon PEX14 | 2 | 1 | 1 | 50 | 1 | 50 | * |
| 293 T-Rex | Exon PEX14 | 34 | 28 | 6 | 18 | 1 | 3 | |
| 4T1 | Exon Car9 | 6 | 3 | 3 | 50 | 3 | 50 | ** |
| E14 mESC | Enhancer Hba R3 | 28 | 16 | 12 | 43 | 3 | 11 | |
| E14 mESC | Enhancer Hba R4 | 62 | 53 | 9 | 15 | 2 | 3 | |
| E14 mESC | Enhancer Hba Rm | 27 | 17 | 10 | 26 | 5 | 19 | |
| HeLa | Exon SCL38A2 | 23 | 17 | 6 | 26 | 2 | 9 | |
| HeLa | Exon SMPD1 | 150 | 147 | 3 | 2 | 4 | 3 | |
| HT29-mTX-E12 | Exon WFDC2 | 20 | 18 | 2 | 10 | 2 | 10 | |
| CTR M3 36S hiPSC | Exon CLU | 80 | 80 | 0 | 0 | 0 | 0 | *** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blayney, J.; Foster, E.M.; Jagielowicz, M.; Kreuzer, M.; Morotti, M.; Reglinski, K.; Xiao, J.H.; Hublitz, P. Unexpectedly High Levels of Inverted Re-Insertions Using Paired sgRNAs for Genomic Deletions. Methods Protoc. 2020, 3, 53. https://doi.org/10.3390/mps3030053
Blayney J, Foster EM, Jagielowicz M, Kreuzer M, Morotti M, Reglinski K, Xiao JH, Hublitz P. Unexpectedly High Levels of Inverted Re-Insertions Using Paired sgRNAs for Genomic Deletions. Methods and Protocols. 2020; 3(3):53. https://doi.org/10.3390/mps3030053
Chicago/Turabian StyleBlayney, Joseph, Evangeline M. Foster, Marta Jagielowicz, Mira Kreuzer, Matteo Morotti, Katharina Reglinski, Julie Huiyuan Xiao, and Philip Hublitz. 2020. "Unexpectedly High Levels of Inverted Re-Insertions Using Paired sgRNAs for Genomic Deletions" Methods and Protocols 3, no. 3: 53. https://doi.org/10.3390/mps3030053
APA StyleBlayney, J., Foster, E. M., Jagielowicz, M., Kreuzer, M., Morotti, M., Reglinski, K., Xiao, J. H., & Hublitz, P. (2020). Unexpectedly High Levels of Inverted Re-Insertions Using Paired sgRNAs for Genomic Deletions. Methods and Protocols, 3(3), 53. https://doi.org/10.3390/mps3030053