Analysis of GPI-Anchored Receptor Distribution and Dynamics in Live Cells by Tag-Mediated Enzymatic Labeling and FRET

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Plasmids
- R-NcoI-T-cad
- 5’-GATCTCCATGGAGATCTGTGATATTCGTCATGG-3’;
- F-SphI-T-cad
- 5’-GCATGTGCATGCCAGGGTACAAGTGTGCTC-3’;
- F-NcoI-YFP
- 5’-CTGTCACCATGGAGTTAGCAAAGGCGAGGAG-3’;
- R-SphI-YFP
- 5’-GTATATGCATGCTCGCGGTACAGCTCGTCCAT-3’;
- F-NcoI-mTFP1
- 5’-CTGTCACCATGGAGTGAGCAAGGGCGAGGAG-3’;
- R-HinfI-mTFP1
- 5’-TCCATGGAGGAGGACTCCTTCAG-3’;
- F-HinfI-mTFP1
- 5’-GAGGAGGACTCCTTCATCTACGA-3’;
- R-SphI-mTFP1
- 5’-GTATATGCATGCTCCTTGTACAGCTCGTCCATGCC-3’.
2.2. Transformation of Competent E. Coli Cells and the Isolation of Plasmid DNA
2.3. Transfection of Eukaryotic Cells and Selection of Clones
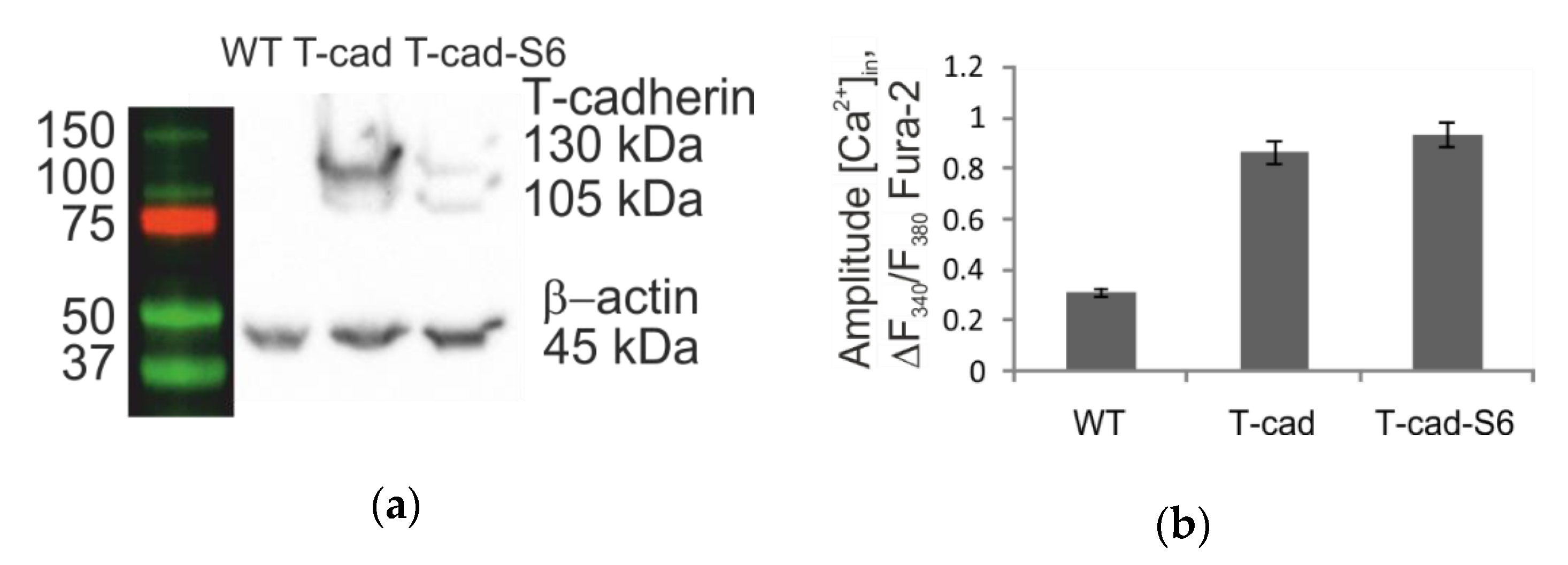
2.4. Comparison of the Molecular Weight, Subcellular Localization, and Functional Activity of the Recombinant Protein
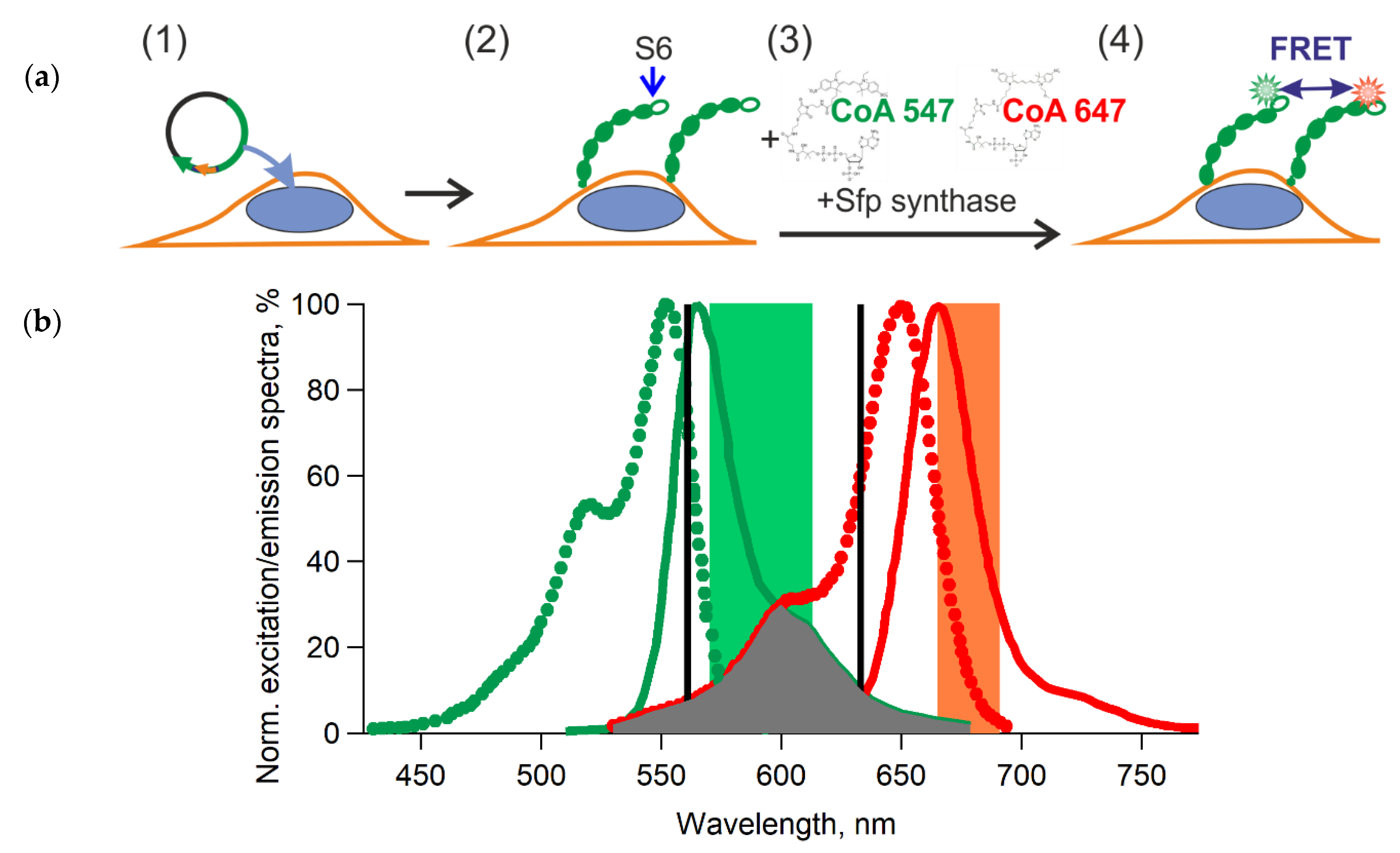
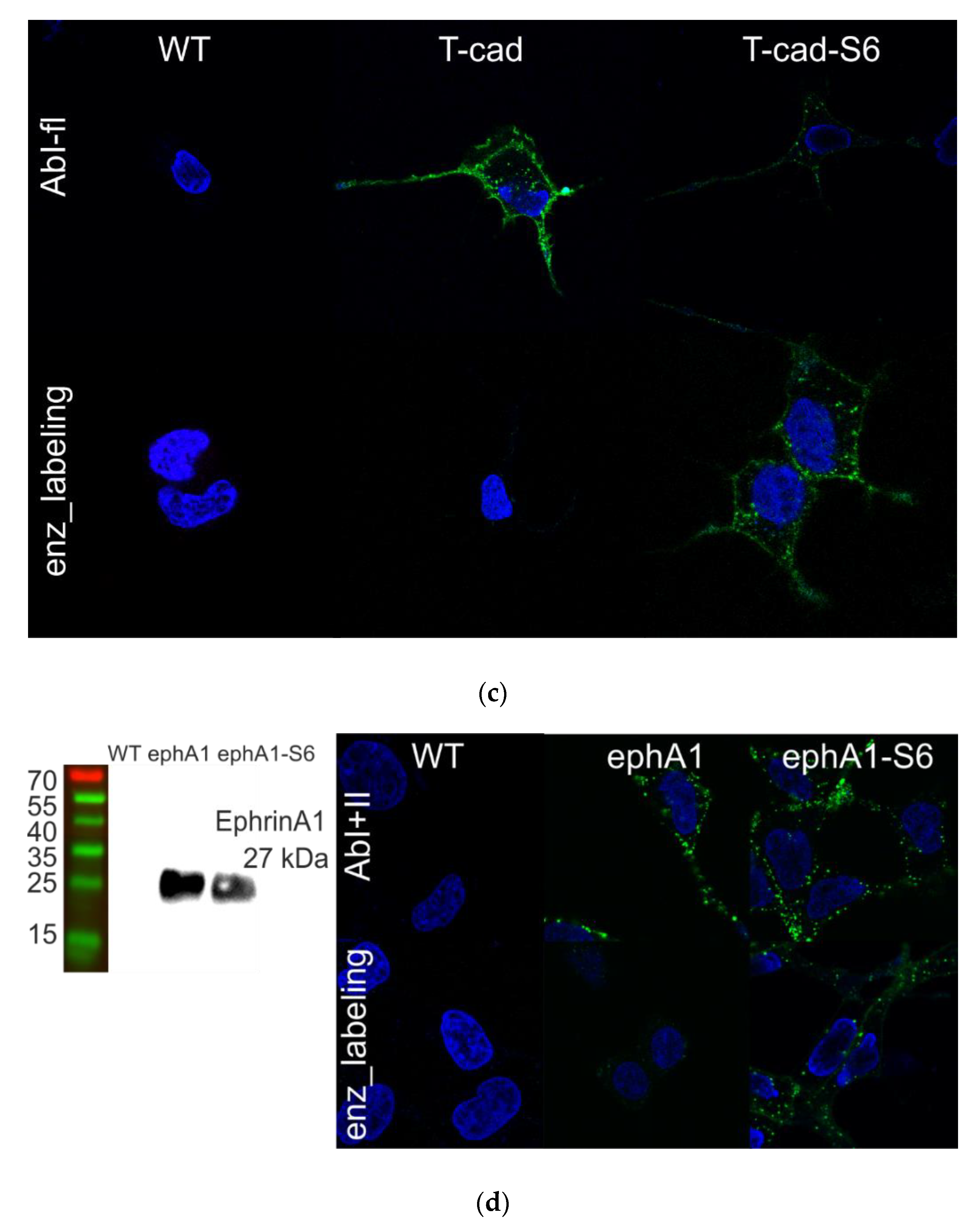
2.5. Enzymatic Labeling of GPI-Anchored Proteins
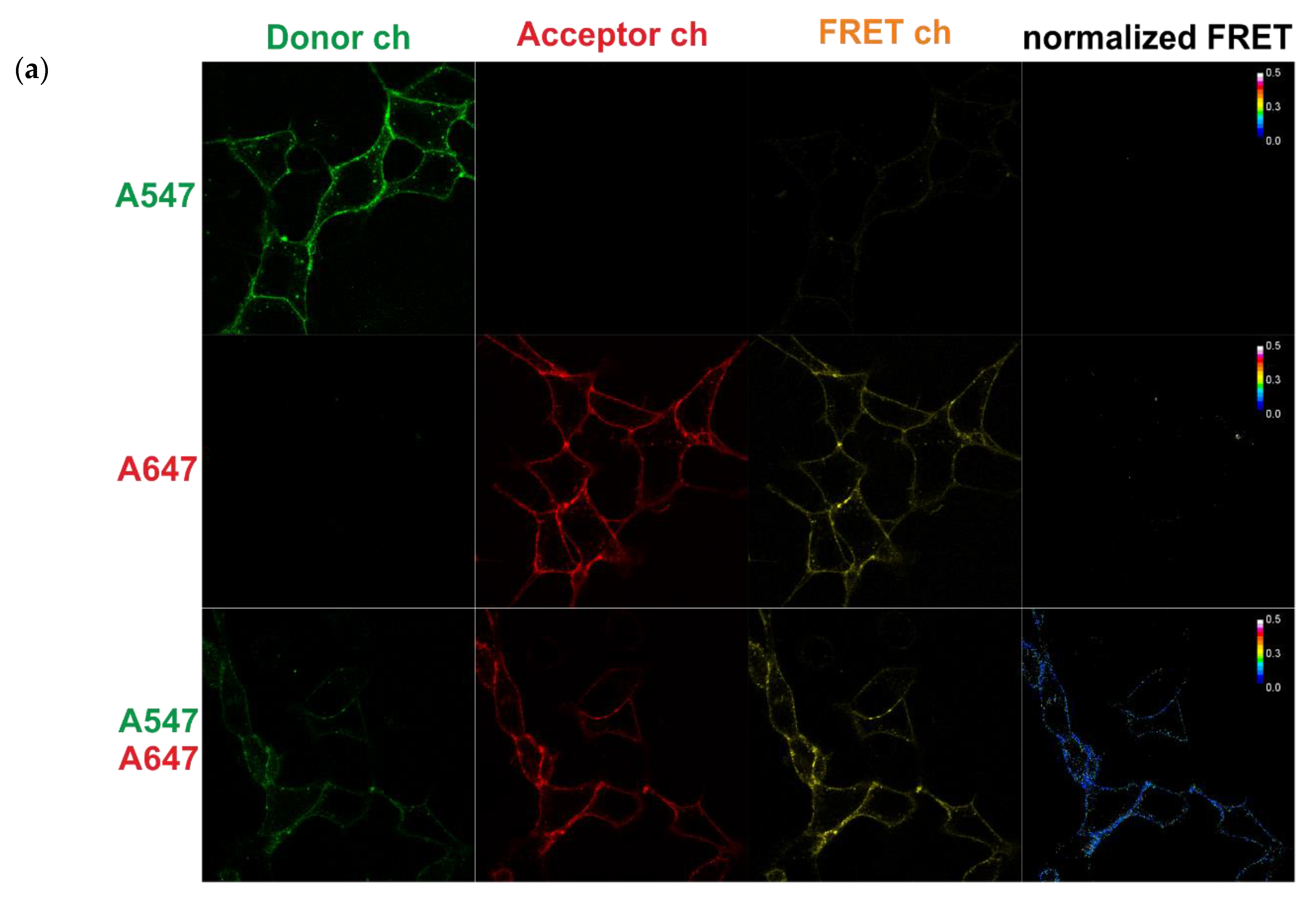
2.6. FRET Measurements Using Confocal Microscopy
2.6.1. Acceptor Photobleaching
2.6.2. Sensitized Acceptor Emission (Confocal Microscopy)
- the control of autofluorescence: blank cells with their proteins of interest (POIs) tagged with S6 peptide without dyes;
- the control of nonspecific binding dyes (the quality of washing step): cells expressing POIs without a tag with the donor + acceptor dyes;
- the control to calculate donor spectral bleed-through: cells expressing POIs tagged with the S6 peptide with donor dye only; and,
- the control to calculate acceptor spectral bleed-through: cells expressing POIs tagged with the S6 peptide with acceptor dye only.
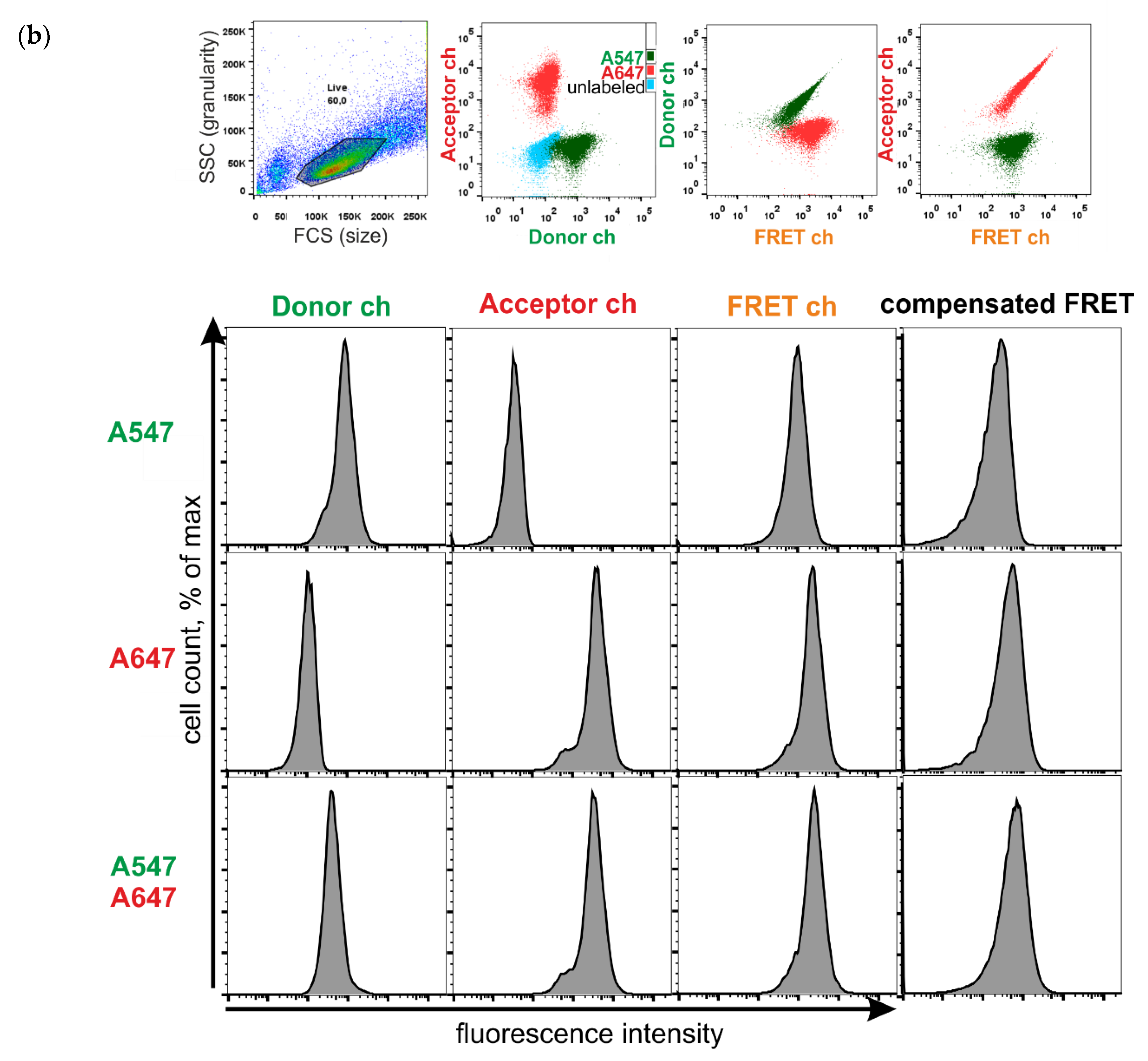
2.7. FRET Measurements Using Flow Cytometry
- Donor ch
- ex 561 em 585/15
- FRET ch
- ex 561 em 670/30
- Acceptor ch
- ex 640 em 670/30
2.8. Kinetics of FRET (Confocal Microscopy and Flow Cytometry)
3. Results and Discussion
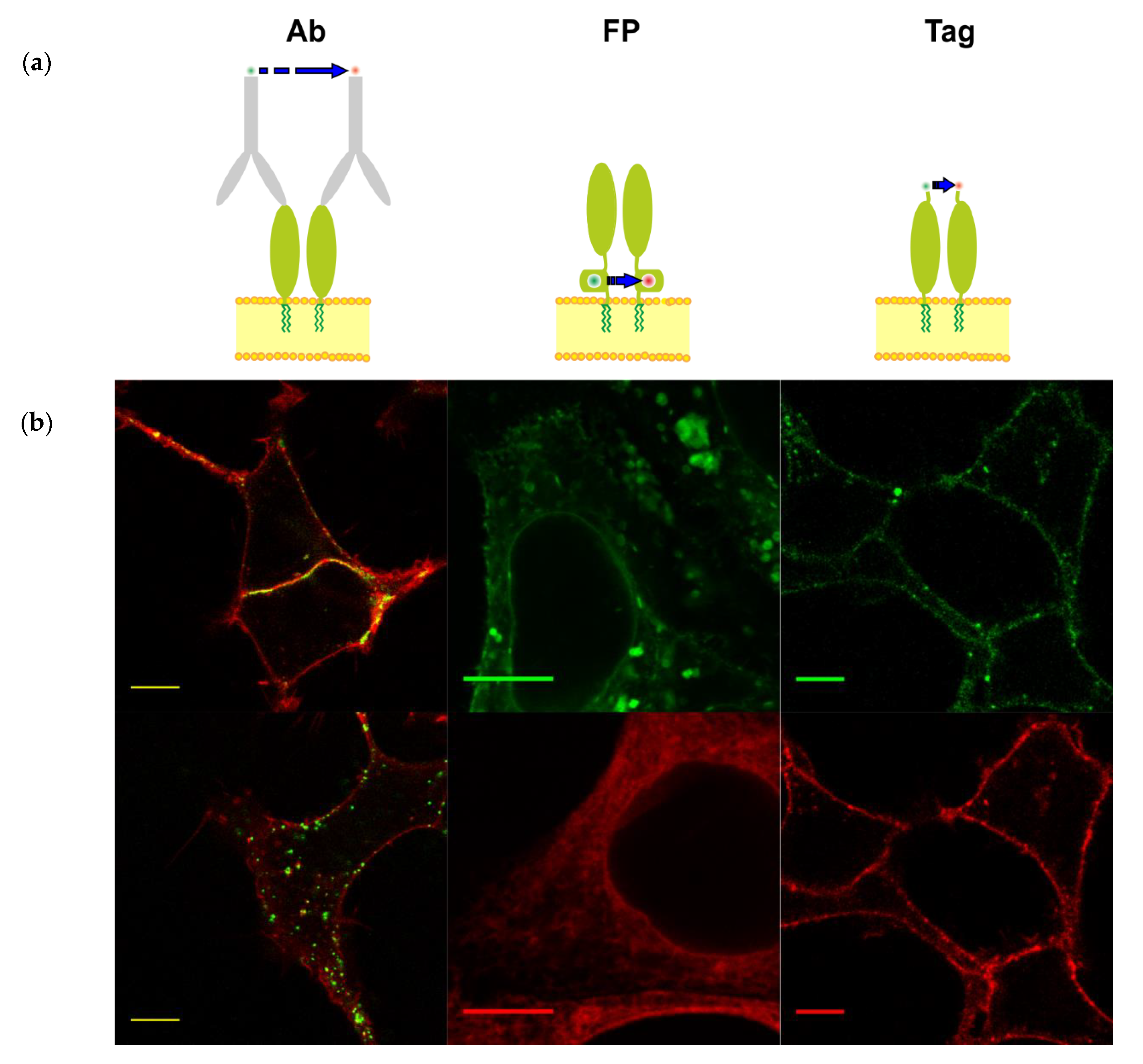
3.1. Different Fluorescent Labeling of GPI-Anchored T-Cadherin
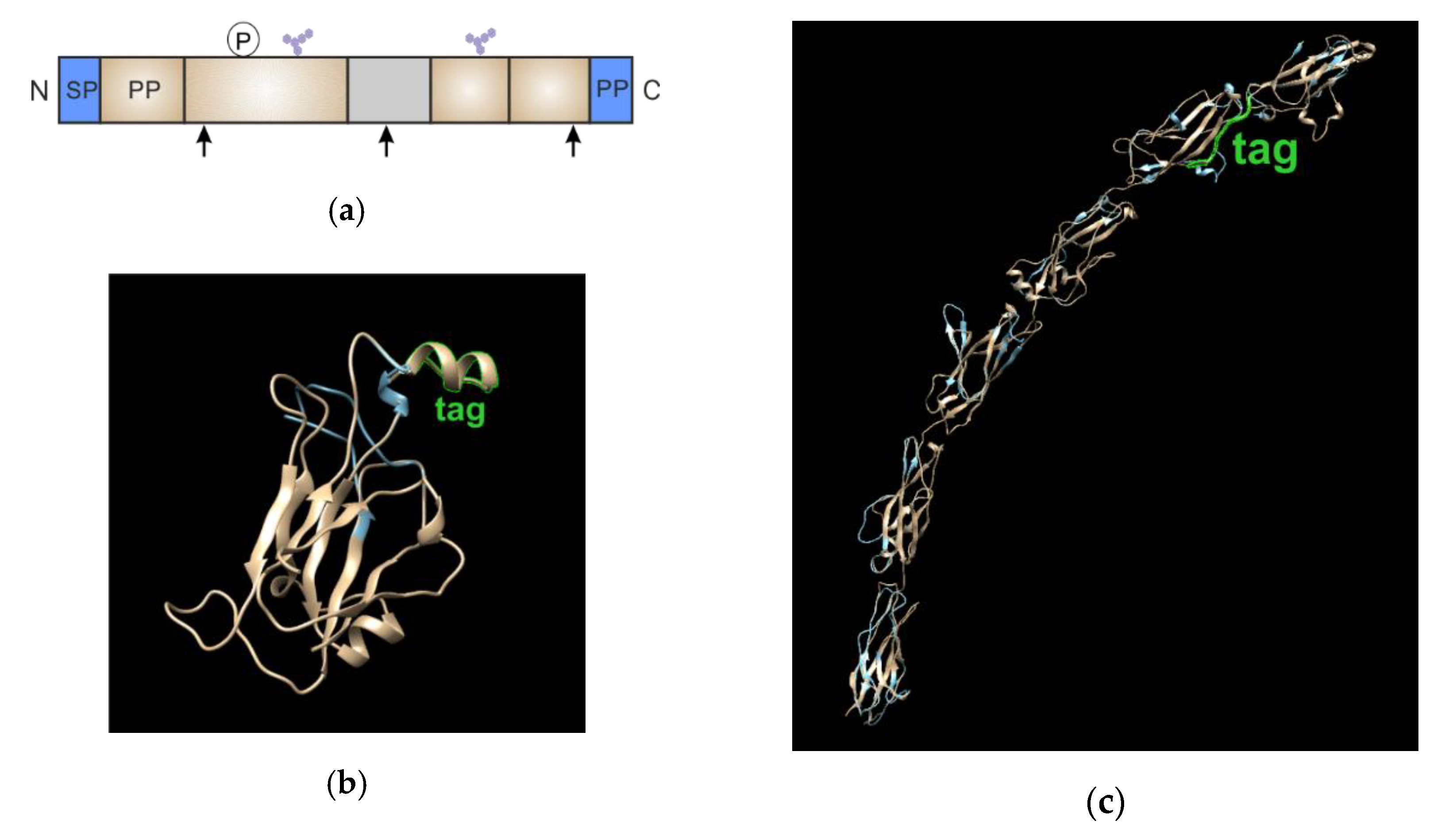
3.2. Validation of Chimeric Proteins with S6-Tag
- 1)
- the inserted tag should be present in the mature protein;
- 2)
- the introduced peptide or polypeptide should not interfere with the enzymes involved in the post-translational modification of the POI (attachment of the GPI anchor, glycosylation, proteolysis, etc.);
- 3)
- modification should not affect the structure of the extracellular domains and ligand-binding sites; and,
- 4)
- the label should be positioned as close as possible to the part of the protein participating in dimerization to maximize the FRET signal.
3.3. FRET between Labeled Molecules on the Cell Surface
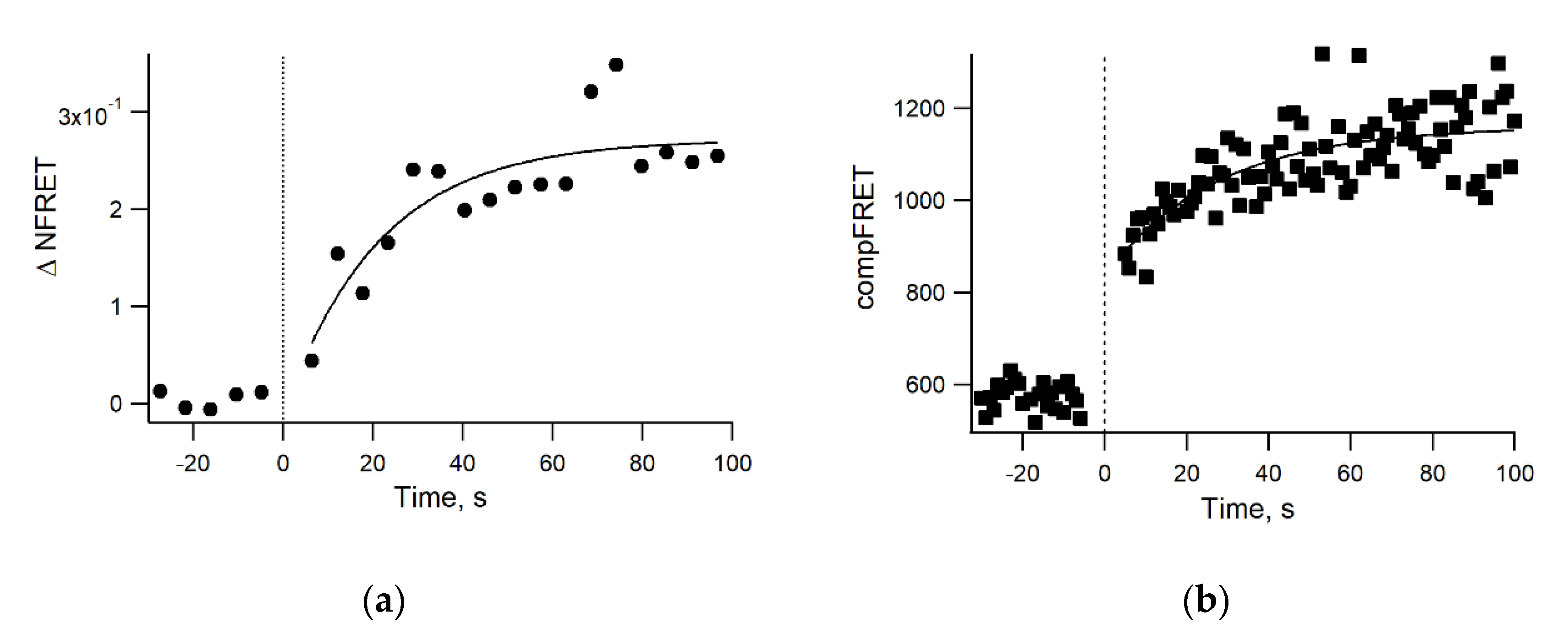
3.4. Real-Time Kinetics of FRET
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1417–1429. [Google Scholar] [CrossRef]
- Sharma, P.; Varma, R.; Sarasij, R.C.; Gousset, K.; Krishnamoorthy, G.; Rao, M.; Mayor, S. Nanoscale Organization of Multiple GPI-Anchored Proteins in Living Cell Membranes. Cell 2004, 116, 577–589. [Google Scholar] [CrossRef]
- Suzuki, K.G.; Kasai, R.S.; Hirosawa, K.M.; Nemoto, Y.L.; Ishibashi, M.; Miwa, Y.; Fujiwara, T.K.; Kusumi, A. Transient GPI-anchored protein homodimers are units for raft organization and function. Nat. Chem. Biol. 2012, 8, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.G.; Fujiwara, T.K.; Sanematsu, F.; Iino, R.; Edidin, M.; Kusumi, A. GPI-anchored receptor clusters transiently recruit Lyn and G alpha for temporary cluster immobilization and Lyn activation: Single-molecule tracking study 1. J. Cell Biol. 2007, 177, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Sevcsik, E.; Brameshuber, M.; Folser, M.; Weghuber, J.; Honigmann, A.; Schutz, G.J. GPI-anchored proteins do not reside in ordered domains in the live cell plasma membrane. Nat. Commun. 2015, 6, 6969. [Google Scholar] [CrossRef]
- Lebreton, S.; Zurzolo, C.; Paladino, S. Organization of GPI-anchored proteins at the cell surface and its physiopathological relevance. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 403–419. [Google Scholar] [CrossRef]
- Kusumi, A.; Fujiwara, T.K.; Morone, N.; Yoshida, K.J.; Chadda, R.; Xie, M.; Kasai, R.S.; Suzuki, K.G. Membrane mechanisms for signal transduction: The coupling of the meso-scale raft domains to membrane-skeleton-induced compartments and dynamic protein complexes. Semin. Cell Dev. Biol. 2012, 23, 126–144. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Edidin, M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 A using imaging fluorescence resonance energy transfer. J. Cell Biol. 1998, 142, 69–84. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef]
- Rao, M.; Mayor, S. Use of Forster’s resonance energy transfer microscopy to study lipid rafts. Biochim. Biophys. Acta 2005, 1746, 221–233. [Google Scholar] [CrossRef]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wallrabe, H.; Seo, S.A.; Periasamy, A. FRET microscopy in 2010: The legacy of Theodor Forster on the 100th anniversary of his birth. Chemphyschem A Eur. J. Chem. Phys. Phys. Chem. 2011, 12, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szollosi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef] [PubMed]
- Szollosi, J.; Vereb, G.; Nagy, P. The flow of events: How the sequence of molecular interactions is seen by the latest, user-friendly high throughput flow cytometric FRET. Cytom. A 2016, 89, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Denzel, M.S.; Scimia, M.C.; Zumstein, P.M.; Walsh, K.; Ruiz-Lozano, P.; Ranscht, B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J. Clin. Investig. 2010, 120, 4342–4352. [Google Scholar] [CrossRef] [PubMed]
- Parker-Duffen, J.L.; Nakamura, K.; Silver, M.; Kikuchi, R.; Tigges, U.; Yoshida, S.; Denzel, M.S.; Ranscht, B.; Walsh, K. T-cadherin Is Essential for Adiponectin-mediated Revascularization. J. Biol. Chem. 2013, 288, 24886–24897. [Google Scholar] [CrossRef]
- Balatskaya, M.N.; Balatskii, A.V.; Sharonov, G.V.; Tkachuk, V.A. T-cadherin as a novel receptor regulating metabolism in the blood vessel and heart cells: From structure to function. J. Evol. Biochem. Phys. 2016, 52, 103–118. [Google Scholar] [CrossRef]
- Fujishima, Y.; Maeda, N.; Matsuda, K.; Masuda, S.; Mori, T.; Fukuda, S.; Sekimoto, R.; Yamaoka, M.; Obata, Y.; Kita, S.; et al. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J. 2017, 31, 1571–1583. [Google Scholar] [CrossRef]
- Obata, Y.; Kita, S.; Koyama, Y.; Fukuda, S.; Takeda, H.; Takahashi, M.; Fujishima, Y.; Nagao, H.; Masuda, S.; Tanaka, Y.; et al. Adiponectin/T-cadherin system enhances exosome biogenesis and decreases cellular ceramides by exosomal release. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Balatskaya, M.N.; Sharonov, G.V.; Baglay, A.I.; Rubtsov, Y.P.; Tkachuk, V.A. Different spatiotemporal organization of GPI-anchored T-cadherin in response to low-density lipoprotein and adiponectin. Biochim. Biophys. Acta Gen. Subj. 2019, 129414. [Google Scholar] [CrossRef]
- Caiolfa, V.R.; Zamai, M.; Malengo, G.; Andolfo, A.; Madsen, C.D.; Sutin, J.; Digman, M.A.; Gratton, E.; Blasi, F.; Sidenius, N. Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J. Cell Biol. 2007, 179, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Tavares, E.; Macedo, J.A.; Paulo, P.M.; Tavares, C.; Lopes, C.; Melo, E.P. Live-cell FRET imaging reveals clustering of the prion protein at the cell surface induced by infectious prions. Biochim. Biophys. Acta 2014, 1842, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cironi, P.; Lin, A.J.; Xu, Y.; Hrvatin, S.; Golan, D.E.; Silver, P.A.; Walsh, C.T.; Yin, J. Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2007, 2, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Straight, P.D.; McLoughlin, S.M.; Zhou, Z.; Lin, A.J.; Golan, D.E.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA 2005, 102, 15815–15820. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ciatto, C.; Bahna, F.; Zampieri, N.; VanSteenhouse, H.C.; Katsamba, P.S.; Ahlsen, G.; Harrison, O.J.; Brasch, J.; Jin, X.; Posy, S.; et al. T-cadherin structures reveal a novel adhesive binding mechanism. Nat. Struct. Mol. Biol. 2010, 17, 339–347. [Google Scholar] [CrossRef]
- Nekrasova, O.V.; Sharonov, G.V.; Tikhonov, R.V.; Kolosov, P.M.; Astapova, M.V.; Yakimov, S.A.; Tagvey, A.I.; Korchagina, A.A.; Bocharova, O.V.; Wulfson, A.N.; et al. Receptor-binding domain of ephrin-A1: Production in bacterial expression system and activity. Biochemistry 2012, 77, 1387–1394. [Google Scholar] [CrossRef]
- Himanen, J.P.; Goldgur, Y.; Miao, H.; Myshkin, E.; Guo, H.; Buck, M.; Nguyen, M.; Rajashankar, K.R.; Wang, B.; Nikolov, D.B. Ligand recognition by A-class Eph receptors: Crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009, 10, 722–728. [Google Scholar] [CrossRef]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef]
- Yin, J.; Lin, A.J.; Golan, D.E.; Walsh, C.T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 2006, 1, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, L.; Buswell, J.; Considine, N.; Ghosh, I.; Masharina, A.; Noren, C.; Xu, M.Q. Fluorescent site-specific labeling of Escherichia coli expressed proteins with Sfp phosphopantetheinyl transferase. Methods Mol. Biol. 2011, 705, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. (Ed.) Principles of Fluorescence Spectroscopy, 2nd ed.; Springer US: Boston, MA, USA, 1999. [Google Scholar] [CrossRef]
- Shapiro, L.; Fannon, A.M.; Kwong, P.D.; Thompson, A.; Lehmann, M.S.; Grubel, G.; Legrand, J.F.; Als-Nielsen, J.; Colman, D.R.; Hendrickson, W.A. Structural basis of cell-cell adhesion by cadherins. Nature 1995, 374, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Berney, C.; Danuser, G. FRET or no FRET: A quantitative comparison. Biophys. J. 2003, 84, 3992–4010. [Google Scholar] [CrossRef]
- Feige, J.N.; Sage, D.; Wahli, W.; Desvergne, B.; Gelman, L. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc. Res. Tech. 2005, 68, 51–58. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Mozgovaja, M.N.; Astapova, M.V.; Kolosov, P.M.; Arseniev, A.S.; Feofanov, A.V. Study of EphA2 dimerization and clusterization in living cells using sensitized acceptor emission in FRET pair. In Current Microscopy Contributions to Advances in Science and Technology; Méndez-Vilas, A., Ed.; FORMATEX: Badajoz, Spain, 2012. [Google Scholar]
- Xia, Z.; Liu, Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001, 81, 2395–2402. [Google Scholar] [CrossRef]
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar] [CrossRef]
- Roederer, M. Compensation in flow cytometry. Curr. Protoc. Cytom. 2002, 22, 1–14. [Google Scholar] [CrossRef]
- Tron, L.; Szollosi, J.; Damjanovich, S.; Helliwell, S.H.; Arndt-Jovin, D.J.; Jovin, T.M. Flow cytometric measurement of fluorescence resonance energy transfer on cell surfaces. Quantitative evaluation of the transfer efficiency on a cell-by-cell basis. Biophys. J. 1984, 45, 939–946. [Google Scholar] [CrossRef]
- Ujlaky-Nagy, L.; Nagy, P.; Szollosi, J.; Vereb, G. Flow Cytometric FRET Analysis of Protein Interactions. Methods Mol. Biol. 2018, 1678, 393–419. [Google Scholar] [CrossRef]
- Szentesi, G.; Horváth, G.; Bori, I.; Vámosi, G.; Szöllősi, J.; Gáspár, R.; Damjanovich, S.; Jenei, A.; Mátyus, L. Computer program for determining fluorescence resonance energy transfer efficiency from flow cytometric data on a cell-by-cell basis. Comput. Methods Programs Biomed. 2004, 75, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Tyrberg, B.; Miles, P.; Azizian, K.T.; Denzel, M.S.; Nieves, M.L.; Monosov, E.Z.; Levine, F.; Ranscht, B. T-cadherin (Cdh13) in association with pancreatic beta-cell granules contributes to second phase insulin secretion. Islets 2011, 3, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; Nguyen, T.A.; van der Meer, B.W.; Blank, P.S. The impact of heterogeneity and dark acceptor states on FRET: Implications for using fluorescent protein donors and acceptors. PLoS ONE 2012, 7, e49593. [Google Scholar] [CrossRef] [PubMed]
- Devauges, V.; Marquer, C.; Lecart, S.; Cossec, J.C.; Potier, M.C.; Fort, E.; Suhling, K.; Leveque-Fort, S. Homodimerization of amyloid precursor protein at the plasma membrane: A homoFRET study by time-resolved fluorescence anisotropy imaging. PLoS ONE 2012, 7, e44434. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, M.E.; Connolly, B.K.; Gurunathan, K.; Levitus, M. Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J. Phys. Chem. B 2007, 111, 11064–11074. [Google Scholar] [CrossRef]
- Banning, C.; Votteler, J.; Hoffmann, D.; Koppensteiner, H.; Warmer, M.; Reimer, R.; Kirchhoff, F.; Schubert, U.; Hauber, J.; Schindler, M. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS ONE 2010, 5, e9344. [Google Scholar] [CrossRef] [PubMed]
- Szollosi, J.; Damjanovich, S.; Matyus, L. Application of fluorescence resonance energy transfer in the clinical laboratory: Routine and research. Cytometry 1998, 34, 159–179. [Google Scholar] [CrossRef]
- Fukuda, S.; Kita, S.; Obata, Y.; Fujishima, Y.; Nagao, H.; Masuda, S.; Tanaka, Y.; Nishizawa, H.; Funahashi, T.; Takagi, J.; et al. The unique prodomain of T-cadherin plays a key role in adiponectin binding with the essential extracellular cadherin repeats 1 and 2. J. Biol. Chem. 2017. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Crivat, G.; Taraska, J.W. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012, 30, 8–16. [Google Scholar] [CrossRef]
- Lotze, J.; Reinhardt, U.; Seitz, O.; Beck-Sickinger, A.G. Peptide-tags for site-specific protein labelling in vitro and in vivo. Mol. Biosyst. 2016, 12, 1731–1745. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| With Fluorescent Antibodies | Chimeras with Fluorescent Proteins | Enzyme-Mediated Fluorescence Labeling of Small Tags | |
|---|---|---|---|
| Genetic modification of the protein of interest (POI) | No | Yes (two constructs) | Yes (one construct) |
| Site specific labeling of the POI | In some cases, the epitope is unknown | Yes | Yes |
| Possibility to use the same cells for control (only donor or acceptor) experiments | Yes | No | Yes |
| Selective labeling of surface-exposed proteins | Yes | No | Yes |
| Approximate volume of the fluorescent label | 400 nm3 | 30 nm3 | 2 nm3 |
| Approximate molecular weight of the label | 150 kDa | 27 kDa | 2.5 kDa |
| Time for labeling reaction | 15–120 min | 0 min | 15–60 min |
| Control of donor: acceptor ratio | Complicated | Complicated | Yes |
| Artificial dimerization | In some cases, the antibodies stimulate dimerization | In some cases, the FPs form a dimer | Not detected |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balatskaya, M.N.; Baglay, A.I.; Rubtsov, Y.P.; Sharonov, G.V. Analysis of GPI-Anchored Receptor Distribution and Dynamics in Live Cells by Tag-Mediated Enzymatic Labeling and FRET. Methods Protoc. 2020, 3, 33. https://doi.org/10.3390/mps3020033
Balatskaya MN, Baglay AI, Rubtsov YP, Sharonov GV. Analysis of GPI-Anchored Receptor Distribution and Dynamics in Live Cells by Tag-Mediated Enzymatic Labeling and FRET. Methods and Protocols. 2020; 3(2):33. https://doi.org/10.3390/mps3020033
Chicago/Turabian StyleBalatskaya, Maria N., Alexandra I. Baglay, Yury P. Rubtsov, and George V. Sharonov. 2020. "Analysis of GPI-Anchored Receptor Distribution and Dynamics in Live Cells by Tag-Mediated Enzymatic Labeling and FRET" Methods and Protocols 3, no. 2: 33. https://doi.org/10.3390/mps3020033
APA StyleBalatskaya, M. N., Baglay, A. I., Rubtsov, Y. P., & Sharonov, G. V. (2020). Analysis of GPI-Anchored Receptor Distribution and Dynamics in Live Cells by Tag-Mediated Enzymatic Labeling and FRET. Methods and Protocols, 3(2), 33. https://doi.org/10.3390/mps3020033