A Simple and Reliable Protocol for the Preparation and Culturing of Fresh Surgically Resected Human Glioblastoma Tissue


Abstract
1. Introduction
2. Experimental Design
 Ensure ALL materials and equipment are sterile prior to usage.
Ensure ALL materials and equipment are sterile prior to usage.2.1. Materials
2.1.1. Poly-D-Lysine Plate Coating Solution
- Poly-D-lysine hydrobromide powder, 5 mg (Merck, Australia; Cat. no.: P6407)
- Sterile distilled water
2.1.2. Enzymatic Tissue Dissociation Solution
- Papain from papaya latex (Merck, Australia; Cat. no.: P3125)
- Earle’s Balanced Salt Solution (EBSS; Thermo Fisher Scientific, Australia; Cat. no.: 14155063)
2.1.3. Culture Medium
- Minimum Essential Medium, 1X, 500 mL (Thermo Fisher Scientific, Australia; Cat. no.: 10370021)
- D-glucose (Merck, Australia; Cat. no.: G7021)
- L-glutamine (Thermo Fisher Scientific, Australia; Cat. no.: 25030081)
- Penicillin-streptomycin (Thermo Fisher Scientific, Australia; Cat. no.: 15070063)
- Heat-inactivated fetal bovine serum (Thermo Fisher Scientific, Australia; Cat. no.: 10100147)
- Corning® MITO+ Serum Extender (Merck, Australia; Cat. no.: DLW355006)
2.2. Equipment
- Class II biological safety cabinet
- 12-well cell culture plates (Merck, Australia; Cat. no.: SIAL0512)
- 18 mm glass coverslips (Thermo Fisher Scientific, Australia; Cat. no.: CB00180RA020MNT0)
- Paraffin
- Surgical tweezer
- 50 mL syringe (Merck, Australia; Cat. no.: Z683698)
- Syringe filter, 0.2 µm pore (Merck, Australia; Cat. no.: CLS431229)
- 50 mL centrifuge tubes (Merck, Australia; Cat. no.: CLS430828)
- Petri dish (Merck, Australia; Cat. no.: P5481)
- Surgical scalpel
- Pipette Boy (Eppendorf, Australia; Cat. no.: 4430000018)
- 10 mL pipettes (Sterilin, Australia; Cat. no.: 47510)
- Pasteur pipette with rubber bulb
- Bunsen burner
- Water bath set at 37 °C
- Automated cell counter
- Humidified 5% CO2/95% O2 incubator, 37 °C (Panasonic; Model no.: MCO-170AICUV-PE)
3. Procedure
 Ensure ALL experiments are completed under sterile conditions with appropriate aseptic techniques to minimize sample contamination and exposure to human tissue.
Ensure ALL experiments are completed under sterile conditions with appropriate aseptic techniques to minimize sample contamination and exposure to human tissue.3.1. Poly-D-Lysine Plate Coating Time for Completion: 3 Days
- Add 50 mL of autoclaved distilled water into 5 mg stock poly-D-lysine powder using a 50 mL syringe with a 0.2 µm pore syringe filter. Re-cap the stock bottle and shake lightly.
- Transfer one 18 mm glass coverslip per well onto 12-well cell culture plates. NOTE: A single 50 mL poly-D-lysine solution can be used to prepare approximately thirteen poly-D-lysine-coated 12-well cell culture plates.
- Add 300 µL of poly-D-lysine solution per well and leave at room temperature (25 °C) for 2 h.
- Aspirate residual poly-D-lysine solution in each well and leave in sterile conditions at room temperature for 48 h or until the wells have dried.
 PAUSE STEP: Seal plates with paraffin and store at 2–8 °C until usage.
PAUSE STEP: Seal plates with paraffin and store at 2–8 °C until usage.

3.2. Sample Preparation and Tissue Culture. Time for Completion: 75 Min for Sample Preparation. 7 Days to Reach 80% Confluency
- 6.
- Pre-warm 10 mL of EBSS in a 50 mL centrifuge tube and culture medium at 37 °C.
- 7.
- Immediately after collection of the fresh sample, add 200 units of papain from papaya latex into warm EBSS.NOTE: This concentration of papain applies to a sample of approximately 5 mm2. Increase concentration with larger samples.
- 8.
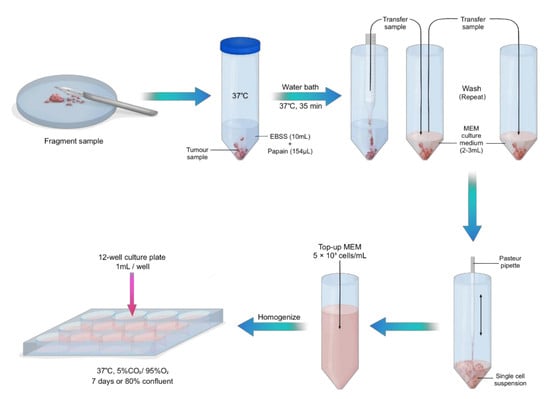
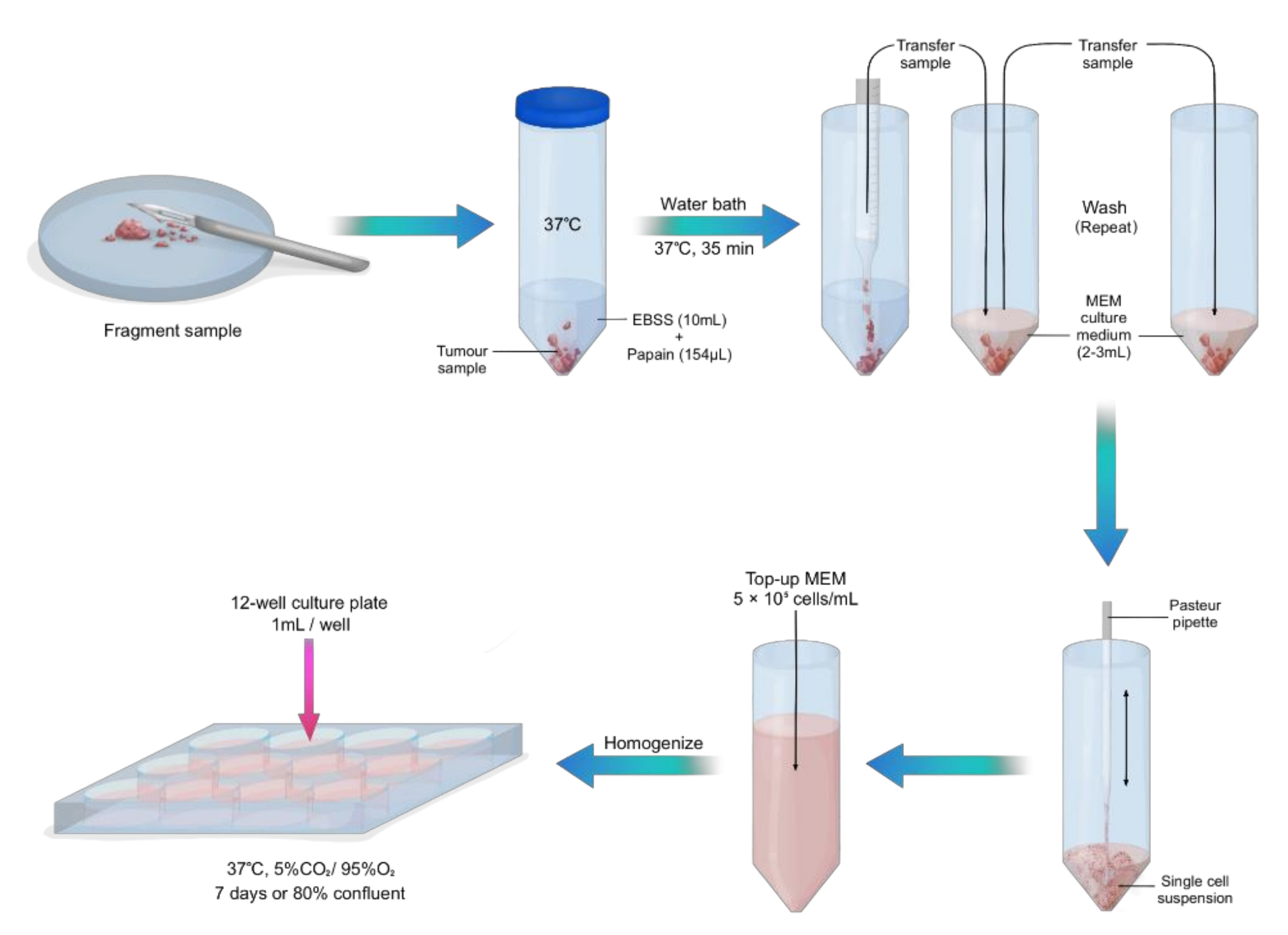
 CRITICAL STEP: Transfer the tumour tissue into a petri dish and gently fragment the tissue with a surgical scalpel (Figure 1).
CRITICAL STEP: Transfer the tumour tissue into a petri dish and gently fragment the tissue with a surgical scalpel (Figure 1).- 9.
- Transfer fragmented sample into the warm EBSS and papain solution and place in a water bath at 37 °C for 40 min.
- 10.
- Gently aspirate sample with a 10 mL pipette and transfer into a new 50 mL centrifuge tube with 3 mL culture medium. Wash in triplicate with culture medium, topping up with new culture medium after each wash.
- 11.
 CRITICAL STEP: With a gentle up and down pipetting motion, use a Pasteur pipette with a rubber bulb to disassociate the cells, creating a single cell suspension.TIP: Soften the edges of the Pasteur pipette with a Bunsen burner to minimize disruption to cellular integrity.
CRITICAL STEP: With a gentle up and down pipetting motion, use a Pasteur pipette with a rubber bulb to disassociate the cells, creating a single cell suspension.TIP: Soften the edges of the Pasteur pipette with a Bunsen burner to minimize disruption to cellular integrity.- 12.
- Count the cells using an automated cell counter and top up accordingly with culture medium to make up 5 × 105 viable (live) cells/mL. Alternatively, a haemocytometer can be used.
- 13.
- Homogenize the cell solution by pipetting up and down and transfer 1 mL per well onto 12-well cell culture plates pre-coated with poly-D-lysine. Maintain cells in a humidified incubator at 37 °C with 5% CO2 for 7 days or until cells are 80% confluent.NOTE: For optimal growth, replace the culture medium every 3–4 days.


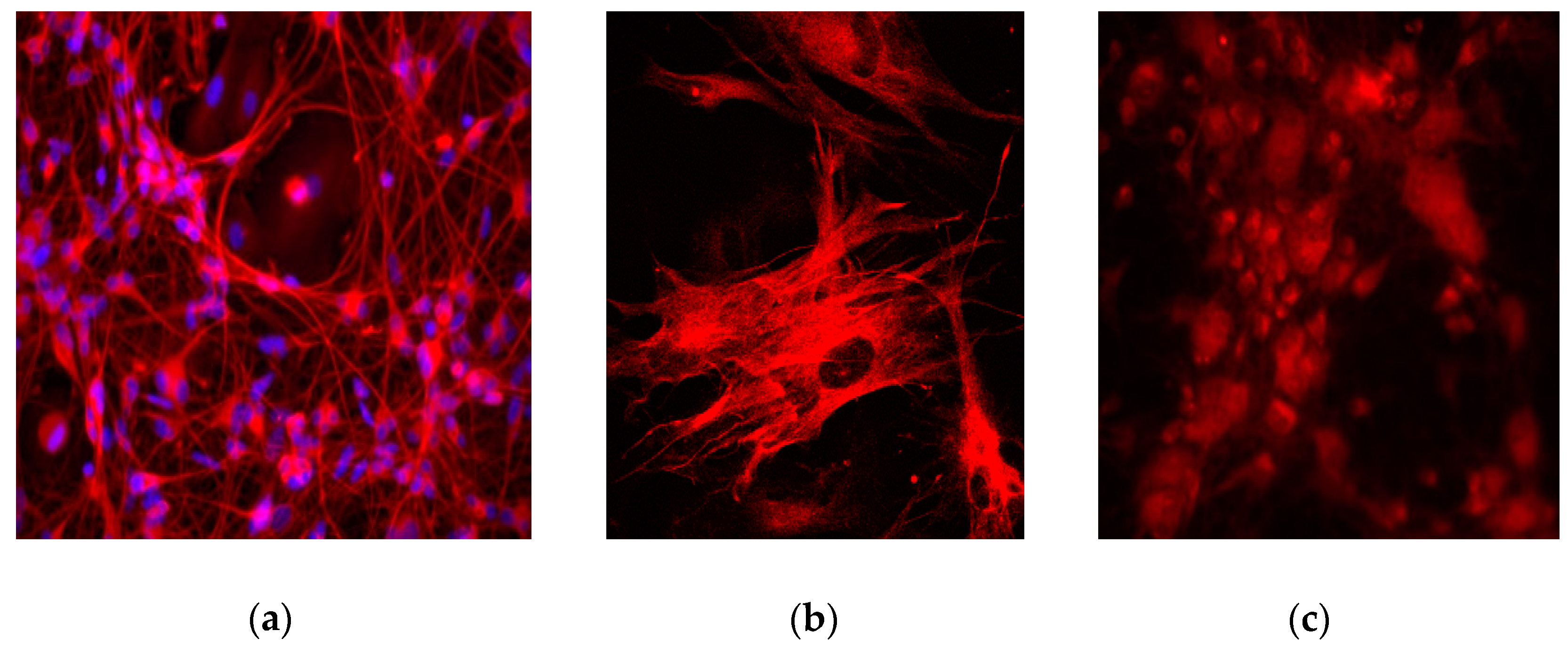
4. Expected Results
Troubleshooting
5. Reagents Setup
5.1. Poly-D-Lysine Plate Coating Solution
- Poly-D-lysine hydrobromide powder, 5 mg
- 50 mL sterile distilled water
5.2. Enzymatic Tissue Dissociation Solution
- 200 units papain from papaya latex (approximately 155 µL in 10 mL EBSS)
- 10 mL EBSS
5.3. Culture Medium
- Minimum Essential Medium, 1 ×, 500 mL
- 1 mM D-glucose
- 2 mM L-glutamine
- 50 units/mL penicillin-streptomycin
- 10% heat-inactivated foetal bovine serum
- Corning® MITO+ Serum Extender (Merck, Australia; Cat. no.: DLW355006)
Author Contributions
Funding
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [PubMed]
- Domingues, P.; González-Tablas, M.; Otero, Á.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Gonçalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G. Roles of purinergic p2x7 receptor in glioma and microglia in brain tumors. Cancer Lett. 2017, 402, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bfgf and egf more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. MCR 2008, 6, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Monif, M.; O’Brien, T.J.; Drummond, K.J.; Reid, C.A.; Liubinas, S.V.; Williams, D.A. P2x7 receptors are a potential novel target for anti-glioma therapies. J. Inflamm. 2014, 11, 25. [Google Scholar] [CrossRef]
- Kan, L.K.; Williams, D.; Drummond, K.; O’Brien, T.; Monif, M. The role of microglia and p2x7 receptors in gliomas. J. Neuroimmunol. 2019, 332, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector t-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived tgf-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.V.; Lipatova, A.V.; Kochetkov, D.V.; Chumakov, P.M. Changes in the sensitivity of human glioblastoma cells to oncolytic enteroviruses induced by passaging. Bull. Russ. State Med Univ. 2018, 2. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin cd11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef] [PubMed]
- Soderquest, K.; Powell, N.; Luci, C.; van Rooijen, N.; Hidalgo, A.; Geissmann, F.; Walzer, T.; Lord, G.M.; Martin-Fontecha, A. Monocytes control natural killer cell differentiation to effector phenotypes. Blood 2011, 117, 4511–4518. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, B.K.A.; Ebaid, H. Expression of cd11b and cd18 on polymorphonuclear neutrophils stimulated with interleukin-2. Cent. Eur. J. Immunol. 2014, 39, 209–215. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Step | Issue | Causes | Suggestions |
|---|---|---|---|
| 11 | Tissue does not easily dissociate when pipetted |
|
|
| 13 | Larger chunks of tissue are present in homogenized cell solution | Tissue was not properly dissociated in Step 11 |
|
| 13 | Cells are not becoming confluent |
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, L.K.; Drummond, K.J.; Hunn, M.; Williams, D.A.; O’Brien, T.J.; Monif, M. A Simple and Reliable Protocol for the Preparation and Culturing of Fresh Surgically Resected Human Glioblastoma Tissue. Methods Protoc. 2020, 3, 11. https://doi.org/10.3390/mps3010011
Kan LK, Drummond KJ, Hunn M, Williams DA, O’Brien TJ, Monif M. A Simple and Reliable Protocol for the Preparation and Culturing of Fresh Surgically Resected Human Glioblastoma Tissue. Methods and Protocols. 2020; 3(1):11. https://doi.org/10.3390/mps3010011
Chicago/Turabian StyleKan, Liyen Katrina, Katharine J Drummond, Martin Hunn, David A Williams, Terence J O’Brien, and Mastura Monif. 2020. "A Simple and Reliable Protocol for the Preparation and Culturing of Fresh Surgically Resected Human Glioblastoma Tissue" Methods and Protocols 3, no. 1: 11. https://doi.org/10.3390/mps3010011
APA StyleKan, L. K., Drummond, K. J., Hunn, M., Williams, D. A., O’Brien, T. J., & Monif, M. (2020). A Simple and Reliable Protocol for the Preparation and Culturing of Fresh Surgically Resected Human Glioblastoma Tissue. Methods and Protocols, 3(1), 11. https://doi.org/10.3390/mps3010011