Optimizing Cell-Free Protein Synthesis for Increased Yield and Activity of Colicins



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Plasmids
2.2. Crude Extract Preparation
2.3. Preparing Linear DNA Template for Cell-Free Colicin Production
2.4. Preparing E3 Immunity Protein
2.5. CFPS Reaction
2.6. Quantifying Colicins Using Radioactive 14C-Leu Assay
2.7. Cell Viability Test
2.8. Statistical Analysis
3. Results and Discussion
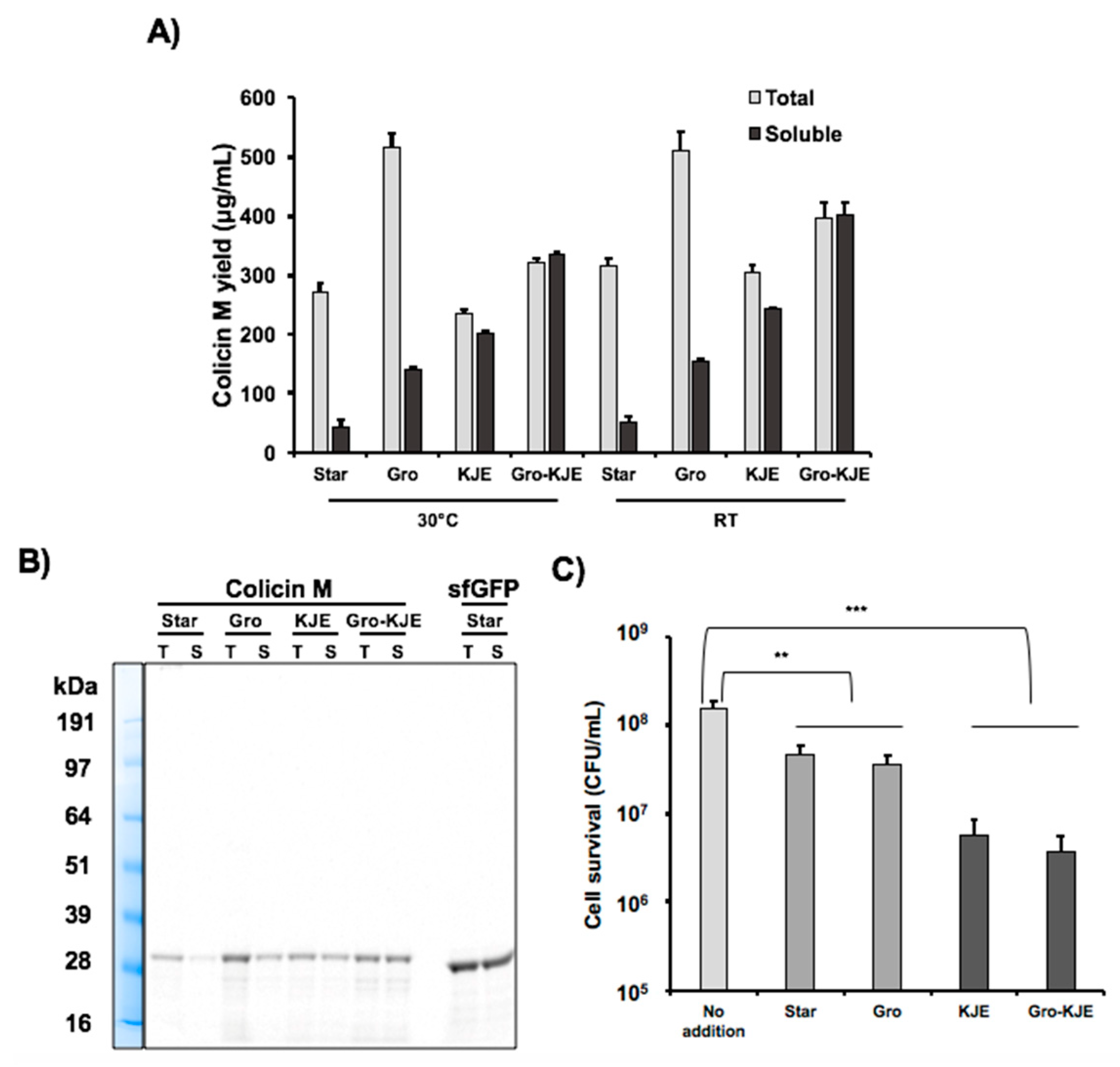
3.1. Enrichment of Cell Extracts with Chaperones Does Not Significantly Affect CFPS Productivity
3.2. Solubility of Colicin M is Increased in the Presence of Chaperones in CFPS
3.3. Increased Colicin M Solubility Enhances Cell-Killing Activity
3.4. Co-Expression of Colicin E3 and Its Immunity Protein Enhances E3 Activity
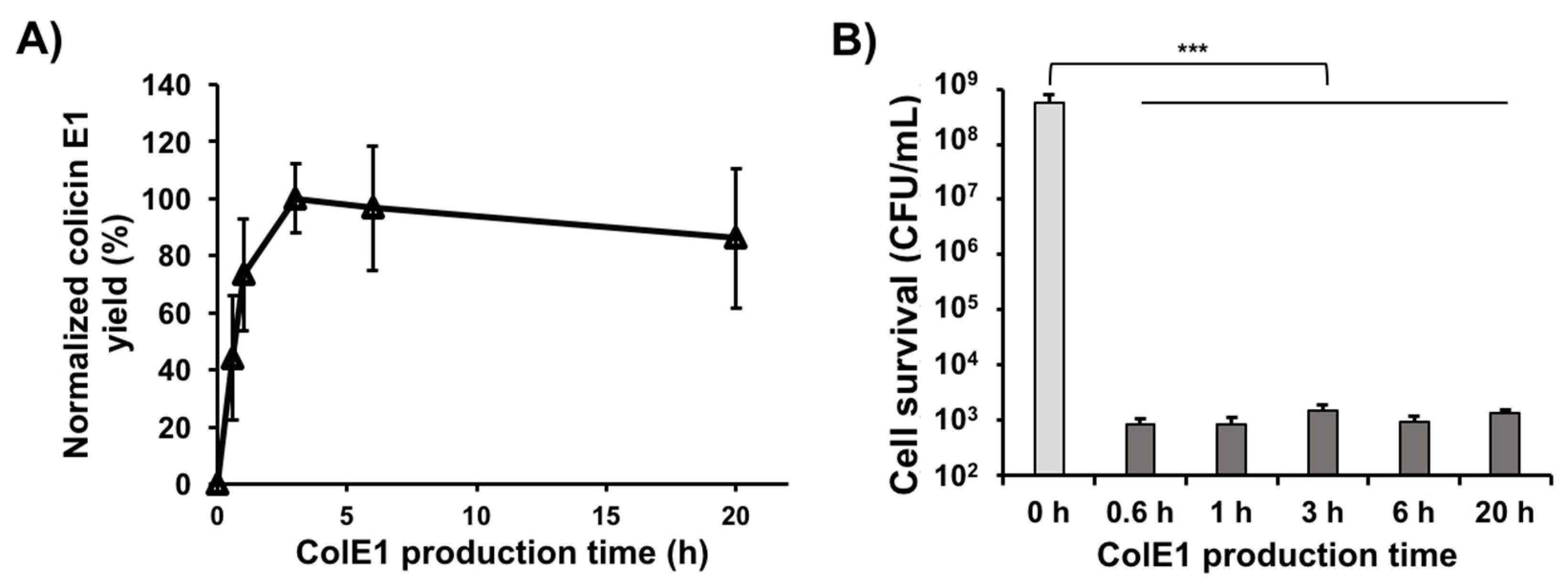
3.5. Colicin E1 Is Rapidly Produced and Remains Stable in CFPS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Deak, D.; Outterson, K.; Powers, J.H.; Kesselheim, A.S. Progress in the fight against multidrug-resistant bacteria? a review of U.S. Food and Drug Administration–approved antibiotics, 2010–2015. Ann. Intern. Med. 2016, 165, 363–372. [Google Scholar] [CrossRef]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef]
- Lee, J.Y.; Bang, D. Challenges in the chemical synthesis of average sized proteins: Sequential vs. convergent ligation of multiple peptide fragments. Biopolymers 2010, 94, 441–447. [Google Scholar] [CrossRef]
- Salehi, A.S.M.; Smith, M.T.; Bennett, A.M.; Williams, J.B.; Pitt, W.G.; Bundy, B.C. Cell-free protein synthesis of a cytotoxic cancer therapeutic: Onconase production and a just-add-water cell-free system. Biotechnol. J. 2016, 11, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.H.C.; Schorch, B.; Boundy, S.; Ffrench-Constant, R.; Kubick, S.; Aktories, K. Cell-free synthesis and characterization of a novel cytotoxic pierisin-like protein from the cabbage butterfly Pieris rapae. Toxicon 2011, 57, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Martemyanov, K.A.; Shirokov, V.A.; Kurnasov, O.V.; Gudkov, A.T.; Spirin, A.S. Cell-free production of biologically active polypeptides: Application to the synthesis of antibacterial peptide cecropin. Protein Expr. Purif. 2001, 21, 456–461. [Google Scholar] [CrossRef]
- Jin, X.; Kightlinger, W.; Kwon, Y.-C.; Hong, S.H. Rapid production and characterization of antimicrobial colicins using Escherichia coli-based cell-free protein synthesis. Synth. Biol. 2018, 3, ysy004. [Google Scholar] [CrossRef]
- Jin, X.; Hong, S.H. Cell-free protein synthesis for producing ‘difficult-to-express’ proteins. Biochem. Eng. J. 2018, 138, 156–164. [Google Scholar] [CrossRef]
- Liu, W.-Q.; Zhang, L.; Chen, M.; Li, J. Cell-free protein synthesis: Recent advances in bacterial extract sources and expanded applications. Biochem. Eng. J. 2019, 141, 182–189. [Google Scholar] [CrossRef]
- Bundy, B.C.; Hunt, J.P.; Jewett, M.C.; Swartz, J.R.; Wood, D.W.; Frey, D.D.; Rao, G. Cell-free biomanufacturing. Curr. Opin. Chem. Eng. 2018, 22, 177–183. [Google Scholar] [CrossRef]
- Cascales, E.; Buchanan, S.K.; Duché, D.; Kleanthous, C.; Lloubès, R.; Postle, K.; Riley, M.; Slatin, S.; Cavard, D. Colicin biology. Microbiol. Mol. Biol. Rev. 2007, 71, 158–229. [Google Scholar] [CrossRef]
- El Ghachi, M.; Bouhss, A.; Barreteau, H.; Touzé, T.; Auger, G.; Blanot, D.; Mengin-Lecreulx, D. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 2006, 281, 22761–22772. [Google Scholar] [CrossRef]
- Zakharov, S.D.; Wang, X.S.; Cramer, W.A. The colicin E1 TolC-binding conformer: Pillar or pore function of TolC in colicin import? Biochemistry 2016, 55, 5084–5094. [Google Scholar] [CrossRef]
- Jakes, K.S.; Finkelstein, A. The colicin Ia receptor, Cir, is also the translocator for colicin Ia. Mol. Microbiol. 2010, 75, 567–578. [Google Scholar] [CrossRef]
- Sharma, O.; Yamashita, E.; Zhalnina, M.V.; Zakharov, S.D.; Datsenko, K.A.; Wanner, B.L.; Cramer, W.A. Structure of the complex of the colicin E2 R-domain and its BtuB receptor. The outer membrane colicin translocon. J. Biol. Chem. 2007, 282, 23163–23170. [Google Scholar] [CrossRef]
- Cramer, W.A.; Sharma, O.; Zakharov, S.D. On mechanisms of colicin import: The outer membrane quandary. Biochem. J. 2018, 475, 3903–3915. [Google Scholar] [CrossRef]
- González-Montalbán, N.; García-Fruitós, E.; Villaverde, A. Recombinant protein solubility—Does more mean better? Nat. Biotechnol. 2007, 25, 718–720. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Niwa, T.; Kanamori, T.; Ueda, T.; Taguchi, H. Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc. Natl. Acad. Sci. USA 2012, 109, 8937–8942. [Google Scholar] [CrossRef]
- Stech, M.; Kubick, S. Cell-free synthesis meets antibody production: A review. Antibodies 2015, 4, 12–33. [Google Scholar] [CrossRef]
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1999, 79, 425–449. [Google Scholar] [CrossRef]
- Oh, I.-S.; Lee, J.-C.; Lee, M.; Chung, J.; Kim, D.-M. Cell-free production of functional antibody fragments. Bioprocess Biosyst. Eng. 2010, 33, 127–132. [Google Scholar] [CrossRef]
- Kang, S.-H.; Kim, D.-M.; Kim, H.-J.; Jun, S.-Y.; Lee, K.-Y.; Kim, H.-J. Cell-free production of aggregation-prone proteins in soluble and active forms. Biotechnol. Prog. 2005, 21, 1412–1419. [Google Scholar] [CrossRef]
- Gopal, G.J.; Kumar, A. Strategies for the production of recombinant protein in Escherichia coli. Protein J. 2013, 32, 419–425. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Chu, H.-S.; Kim, T.-W.; Oh, I.-S.; Choi, C.-Y.; Hahn, G.-H.; Park, C.-G.; Kim, D.-M. Cell-free synthesis of recombinant proteins from PCR-amplified genes at a comparable productivity to that of plasmid-based reactions. Biochem. Biophys. Res. Commun. 2005, 338, 1346–1352. [Google Scholar] [CrossRef]
- Jakes, K.S. Translocation trumps receptor binding in colicin entry into Escherichia coli. Biochem. Soc. Trans. 2012, 40, 1443–1448. [Google Scholar] [CrossRef]
- Kwon, Y.-C.; Jewett, M.C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 2015, 5, 8663. [Google Scholar] [CrossRef]
- Jakes, K.S. The colicin E1 TolC box: Identification of a domain required for colicin E1 cytotoxicity and TolC binding. J. Bacteriol. 2017, 199, e00412-16. [Google Scholar] [CrossRef]
- Hong, S.H.; Ntai, I.; Haimovich, A.D.; Kelleher, N.L.; Isaacs, F.J.; Jewett, M.C. Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific non-standard amino acid incorporation. ACS Synth. Biol. 2014, 3, 398–409. [Google Scholar] [CrossRef]
- Soelaiman, S.; Jakes, K.; Wu, N.; Li, C.; Shoham, M. Crystal structure of colicin E3: Implications for cell entry and ribosome inactivation. Mol. Cell 2001, 8, 1053–1062. [Google Scholar] [CrossRef]
- Swartz, J.R.; Jewett, M.C.; Woodrow, K.A. Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol. 2004, 267, 169–182. [Google Scholar]
- Sharma, O.; Cramer, W.A. Minimum length requirement of the flexible N-terminal translocation subdomain of colicin E3. J. Bacteriol. 2007, 189, 363–368. [Google Scholar] [CrossRef]
- Pédelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Acebrón, S.P.; Fernández-Sáiz, V.; Taneva, S.G.; Moro, F.; Muga, A. DnaJ recruits DnaK to protein aggregates. J. Biol. Chem. 2008, 283, 1381–1390. [Google Scholar] [CrossRef]
- Hayer-Hartl, M.; Bracher, A.; Hartl, F.U. The GroEL-GroES chaperonin machine: A nano-cage for protein folding. Trends Biochem. Sci. 2016, 41, 62–76. [Google Scholar] [CrossRef]
- Choi, G.-H.; Lee, D.-H.; Min, W.-K.; Cho, Y.-J.; Kweon, D.-H.; Son, D.-H.; Park, K.; Seo, J.-H. Cloning, expression, and characterization of single-chain variable fragment antibody against mycotoxin deoxynivalenol in recombinant Escherichia coli. Protein Expr. Purif. 2004, 35, 84–92. [Google Scholar] [CrossRef]
- Vera, A.; González-Montalbán, N.; Arís, A.; Villaverde, A. The conformational quality of insoluble recombinant proteins is enhanced at low growth temperatures. Biotechnol. Bioeng. 2007, 96, 1101–1106. [Google Scholar] [CrossRef]
- Jewett, M.C.; Fritz, B.R.; Timmerman, L.E.; Church, G.M. In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol. Syst. Biol. 2013, 9, 678. [Google Scholar] [CrossRef]
- Ng, C.L.; Lang, K.; Meenan, N.A.G.; Sharma, A.; Kelley, A.C.; Kleanthous, C.; Ramakrishnan, V. Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nat. Struct. Mol. Biol. 2010, 17, 1241–1246. [Google Scholar] [CrossRef][Green Version]
- Salehi, A.S.M.; Yang, S.-O.; Earl, C.C.; Shakalli Tang, M.J.; Porter Hunt, J.; Smith, M.T.; Wood, D.W.; Bundy, B.C. Biosensing estrogenic endocrine disruptors in human blood and urine: A RAPID cell-free protein synthesis approach. Toxicol. Appl. Pharmacol. 2018, 345, 19–25. [Google Scholar] [CrossRef]
- Zakharov, S.D.; Zhalnina, M.V.; Sharma, O.; Cramer, W.A. The colicin E3 outer membrane translocon: Immunity protein release allows interaction of the cytotoxic domain with OmpF porin. Biochemistry 2006, 45, 10199–10207. [Google Scholar] [CrossRef]
- Smith, M.T.; Berkheimer, S.D.; Werner, C.J.; Bundy, B.C. Lyophilized Escherichia coli-based cell-free systems for robust, high-density, long-term storage. Biotechniques 2014, 56, 186–193. [Google Scholar] [CrossRef]
- Hunt, J.P.; Yang, S.O.; Wilding, K.M.; Bundy, B.C. The growing impact of lyophilized cell-free protein expression systems. Bioengineered 2017, 8, 325–330. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef]
- Bidell, M.R.; Palchak, M.; Mohr, J.; Lodise, T.P. Fluoroquinolone and third-generation-cephalosporin resistance among hospitalized patients with urinary tract infections due to Escherichia coli: Do rates vary by hospital characteristics and geographic region? Antimicrob. Agents Chemother. 2016, 60, 3170–3173. [Google Scholar] [CrossRef]
- CIPRO® (Ciprofloxacin Hydrochloride) [Product Information]; Bayer HealthCare Pharmaceutical Inc.: Whippany, NJ, USA, 1987.
- Caschera, F.; Noireaux, V. Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription–translation system. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strains and Plasmids | Genotype/Relevant Characteristics | Source |
|---|---|---|
| Strains | ||
| E. coli K361 | Wild type W3110 strain with StrR | [30] |
| E. coli BL21 Star (DE3) | F− ompT, hsdSB (rB−mB−), gal, dcm, rne131 (DE3) | Invitrogen |
| E. coli TG1 | Strain containing colicin plasmid | [32] |
| Plasmids | ||
| pJL1-sfGFP | KmR, PT7::sfGFP, C-terminal Strep-tag | [33] |
| pJL1-cma | KmR, PT7::cma encoding colicin M | [9] |
| pJL1-E3 imm | KmR, PT7::E3imm encoding E3 immunity | This study |
| pKSJ331 | AmR, ColE1 operon | [32] |
| pKSJ167 | AmR, ColE3 operon | [34] |
| pGro7 | CmR, ParaB::groES-groEL encoding GroES/EL | Takara Bio |
| pKJE7 | CmR, ParaB::dnaK-dnaJ-grpE encoding DnaK/DnaJ/GrpE | Takara Bio |
| pG-KJE8 | CmR, ParaB::dnaK-dnaJ-grpE encoding DnaK/DnaJ/GrpE, PPzt-1::groES-groEL encoding GroES/EL | Takara Bio |
| Extracts | Solubility (%) | |
|---|---|---|
| 30 °C | RT | |
| Star | 16 ± 4 | 16 ± 3 |
| Gro | 27 ± 2 | 30 ± 2 |
| KJE | 86 ± 3 | 80 ± 3 |
| Gro-KJE | 104 ± 2 | 102 ± 9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.; Kightlinger, W.; Hong, S.H. Optimizing Cell-Free Protein Synthesis for Increased Yield and Activity of Colicins. Methods Protoc. 2019, 2, 28. https://doi.org/10.3390/mps2020028
Jin X, Kightlinger W, Hong SH. Optimizing Cell-Free Protein Synthesis for Increased Yield and Activity of Colicins. Methods and Protocols. 2019; 2(2):28. https://doi.org/10.3390/mps2020028
Chicago/Turabian StyleJin, Xing, Weston Kightlinger, and Seok Hoon Hong. 2019. "Optimizing Cell-Free Protein Synthesis for Increased Yield and Activity of Colicins" Methods and Protocols 2, no. 2: 28. https://doi.org/10.3390/mps2020028
APA StyleJin, X., Kightlinger, W., & Hong, S. H. (2019). Optimizing Cell-Free Protein Synthesis for Increased Yield and Activity of Colicins. Methods and Protocols, 2(2), 28. https://doi.org/10.3390/mps2020028