Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools


Abstract
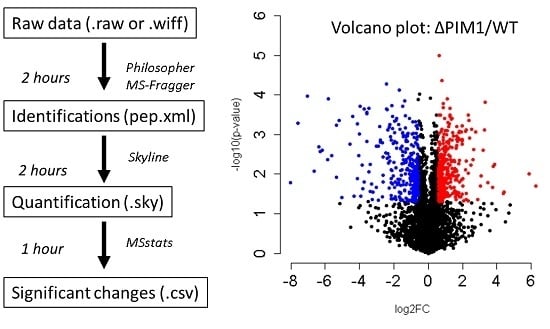
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
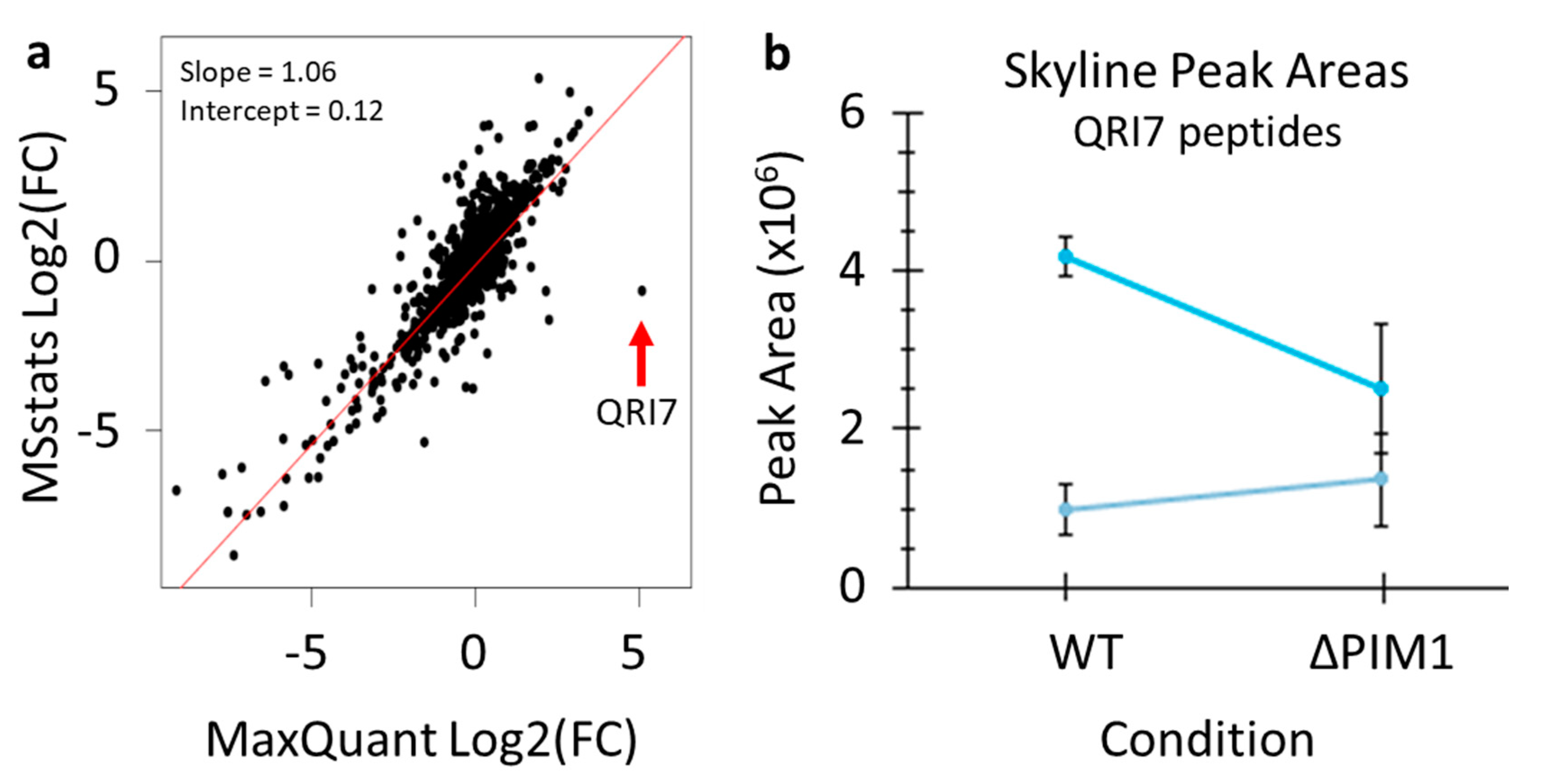{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Design
2.1. Materials
- Raw mass spectrometry data from the DDA proteomics experiment (tutorial data available from ftp://massive.ucsd.edu/MSV000083136/raw/);
- Microsoft .NET Framework 4 (https://www.microsoft.com/en-us/download/details.aspx?id=17851);
- Java runtime environment version 1.8 (https://java.com/en/download/windows-64bit.jsp);
- MSconvert Software, version 3.0.18282 (http://proteowizard.sourceforge.net/download.html);
- FragPipe Software v7.1 (https://github.com/chhh/FragPipe/releases/tag/v7.1);
- MS-Fragger Software, current version (https://bit.ly/2z6dzXa);
- Philosopher executable Build: 201809241411 (https://github.com/prvst/philosopher/releases/tag/20180924 scroll down and select “philosopher_windows_amd64.exe”);
- Skyline Software version 4.2 (https://skyline.ms/project/home/software/Skyline/begin.view).
2.2. Equipment
- A 64-bit computer with Windows 7 or Windows 10 operating system, at least 8 GB of RAM, at least quad-core i5 processor or equivalent, and at least 50 GB of free disk space.
3. Procedure
3.1. Install Required Software and Setup Directories; Time for Completion: 1 Hour
- Follow the instructions on the developer’s websites to install the software programs described under Section 2.1.
- Setup your directories. Make a new directory on your computer’s C drive called “C:\FragPipe_Skyline” and move the philosopher executable file to this folder. Within that folder make the folder “C:\FragPipe_Skyline\data” and move the .RAW files here.
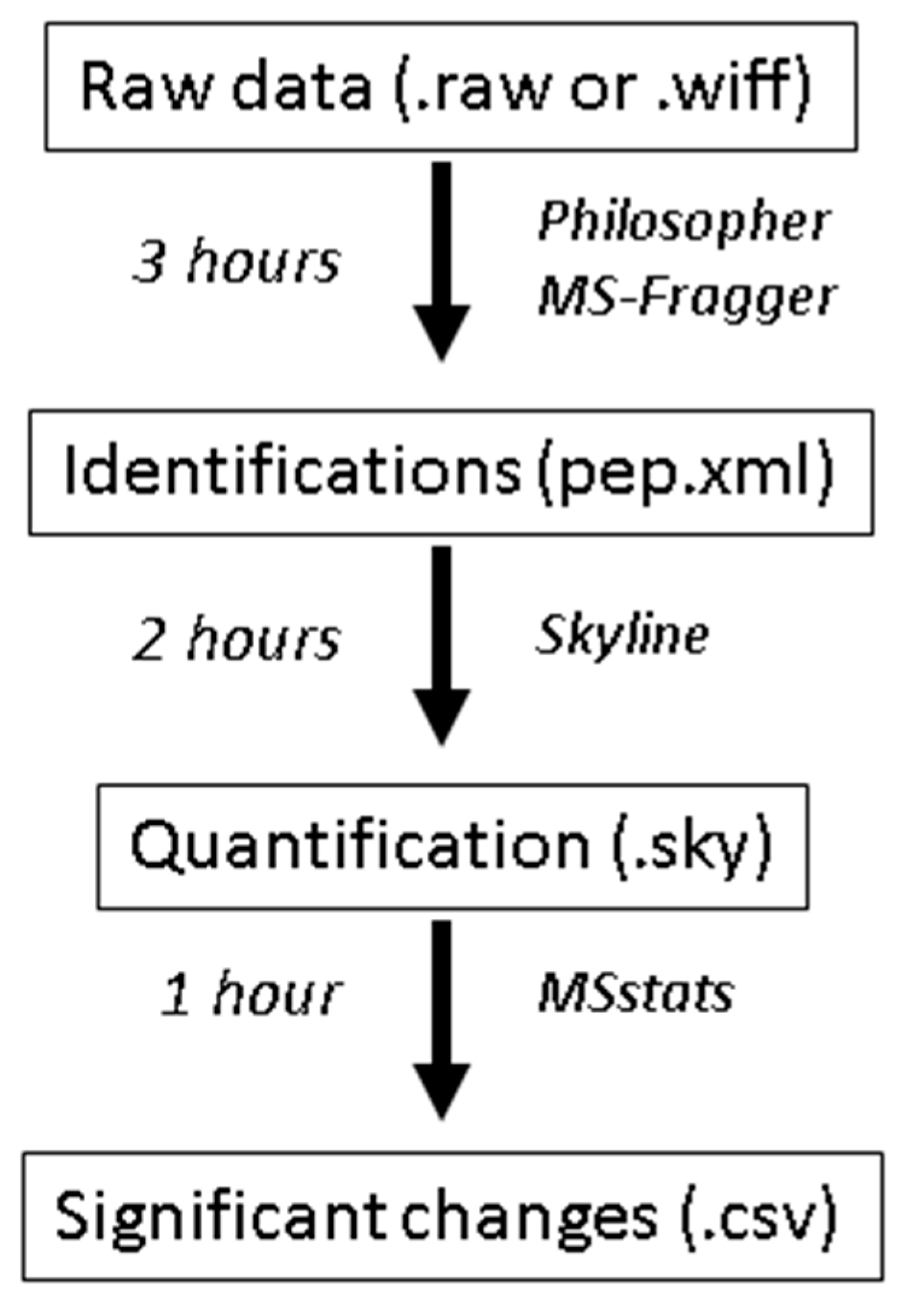
3.2. Identify Peptides Using Database Searching; Time for Completion: 3 Hours
3.2.1. Convert Raw Mass Spectrometry Data to mzXML
- Navigate to your system folder containing the raw mass spectrometry data.
- Select your raw data files (.raw files from Thermo instruments, .wiff files from ABsciex instruments).
- Right click on the selected files and choose “open with MSconvertGUI”.
- In the options box below the output directory, adjust the settings to output format = “mzXML”, Binary encoding precision = “64-bit”, and check the boxes next to “write index”, “use zlib compression”, and “TPP compatibility”.
- In the filter box, select the dropdown box, and choose “peak picking”. Do not change the settings that pop up and click “add”. Your window should look like Figure 1.
- At the bottom right corner, click “start”, and wait for your files to finish converting to mzXML.
3.2.2. Prepare the Organism-Specific Database Using Philosopher
- Navigate to the list of UniProt proteomes in your web browser: https://www.uniprot.org/proteomes/.
- Type the name of your organism into the search box. With the tutorial data, the data is from Saccharomyces cerevisiae (UP000002311).
- Copy the UniProt ID from the column to the left of its name.
- Open a windows command prompt (click the “start” button on the lower left corner, type “cmd” and hit enter.
- Navigate to the location of your philosopher executable using the command “cd [full path to folder]” (Figure 3). For this tutorial, we created a file on the C:\drive with the executables, so we use: cd C:\FragPipe_Skyline\.
- Initialize your philosopher workspace by typing the following (Figure 2):philosopher_windows_amd64.exe workspace –initwhere the first command is the name of your philosopher executable.
- Download your organism database and add contaminants and decoys by typing (Figure 2):philosopher_windows_amd64.exe database --prefix rev_ --contam --id UP000002311where the last text after “—id” is the uniprot identifier for your organism, which for the tutorial data is Saccharomyces cerevisiae (UP000002311). Do not close the command prompt. You will use this again in a subsequent step.
3.2.3. Peptide Identification by Database Search Using FragPipe Interface to MSFragger
- Open FragPipe by double-clicking on Fragpipe.bat.
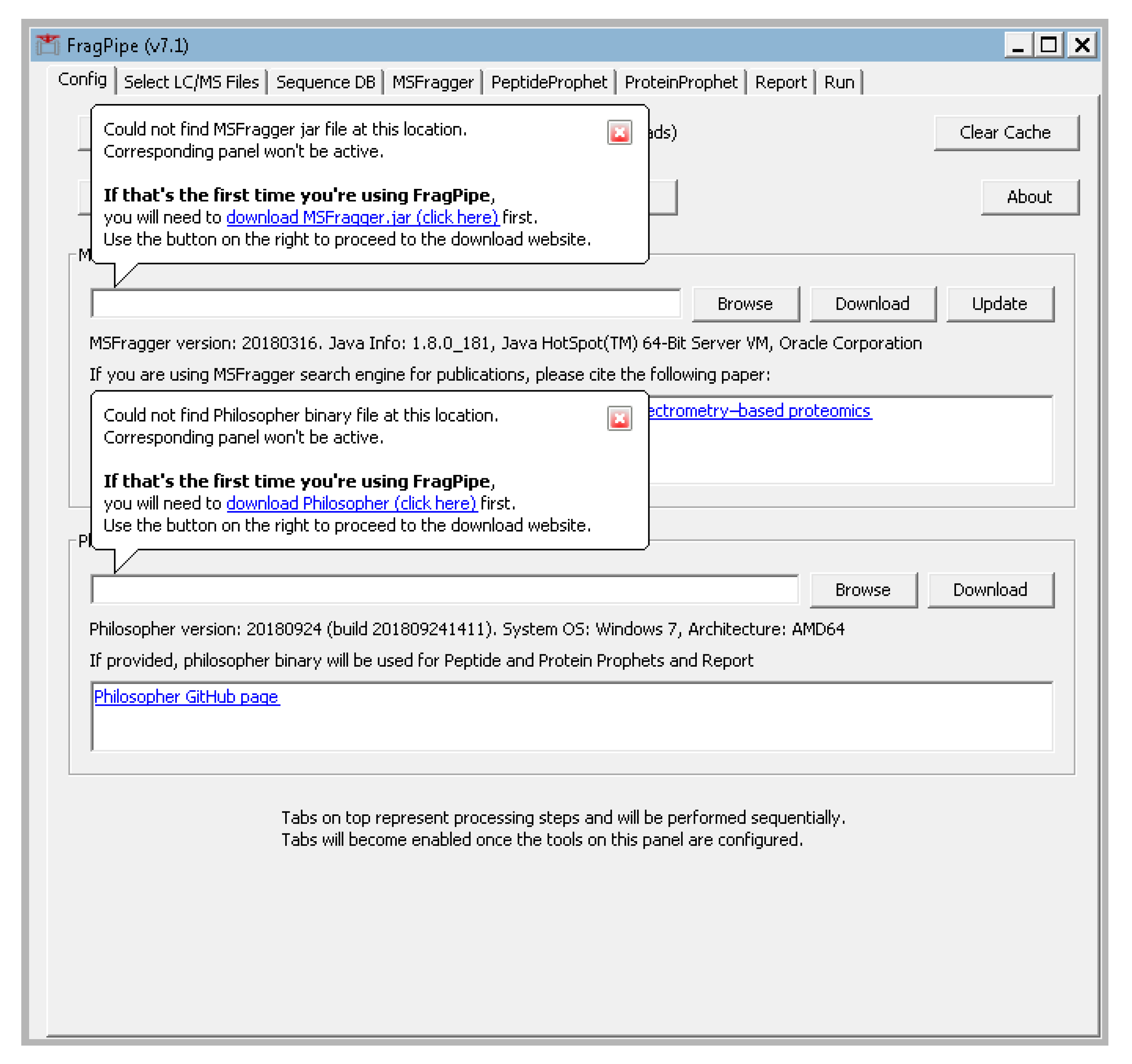
- The FragPipe window should pop up and prompt you for the locations of the MSFragger.jar and philosopher.exe. Click “browse” to navigate to their locations or click the download buttons for links to their download locations (Figure A1 in Appendix A).
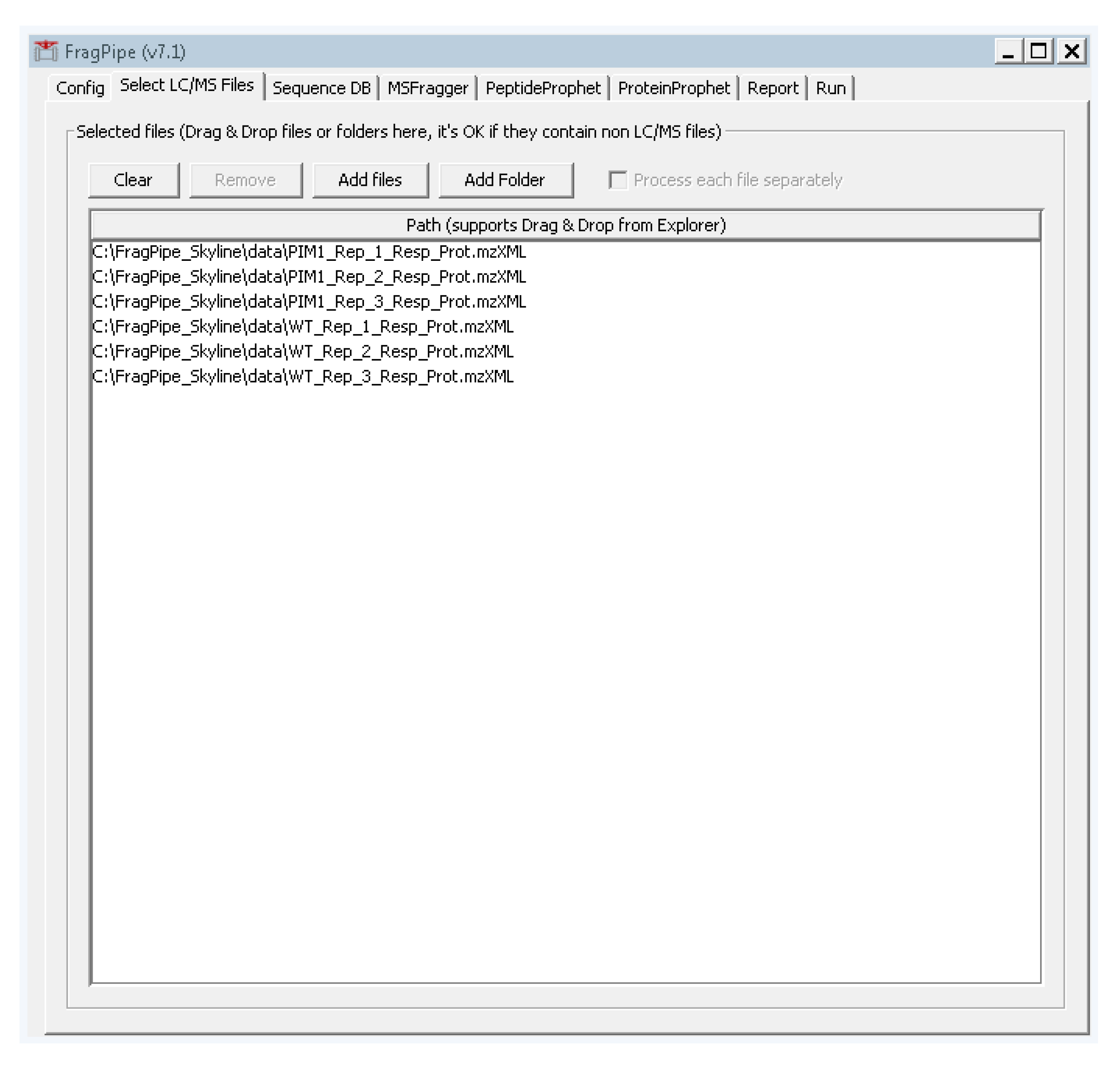
- Select the second tab “Select LC/MS Files” and add the .mzXML files we created in step 2 by either dragging and dropping them into the large white box, or by clicking “add files” and navigating to their location (Figure A2).
- Select the third tab, “sequence DB”, and add the FASTA file we created in step 3 by clicking the “browse” button and navigating to its location (Figure A3).
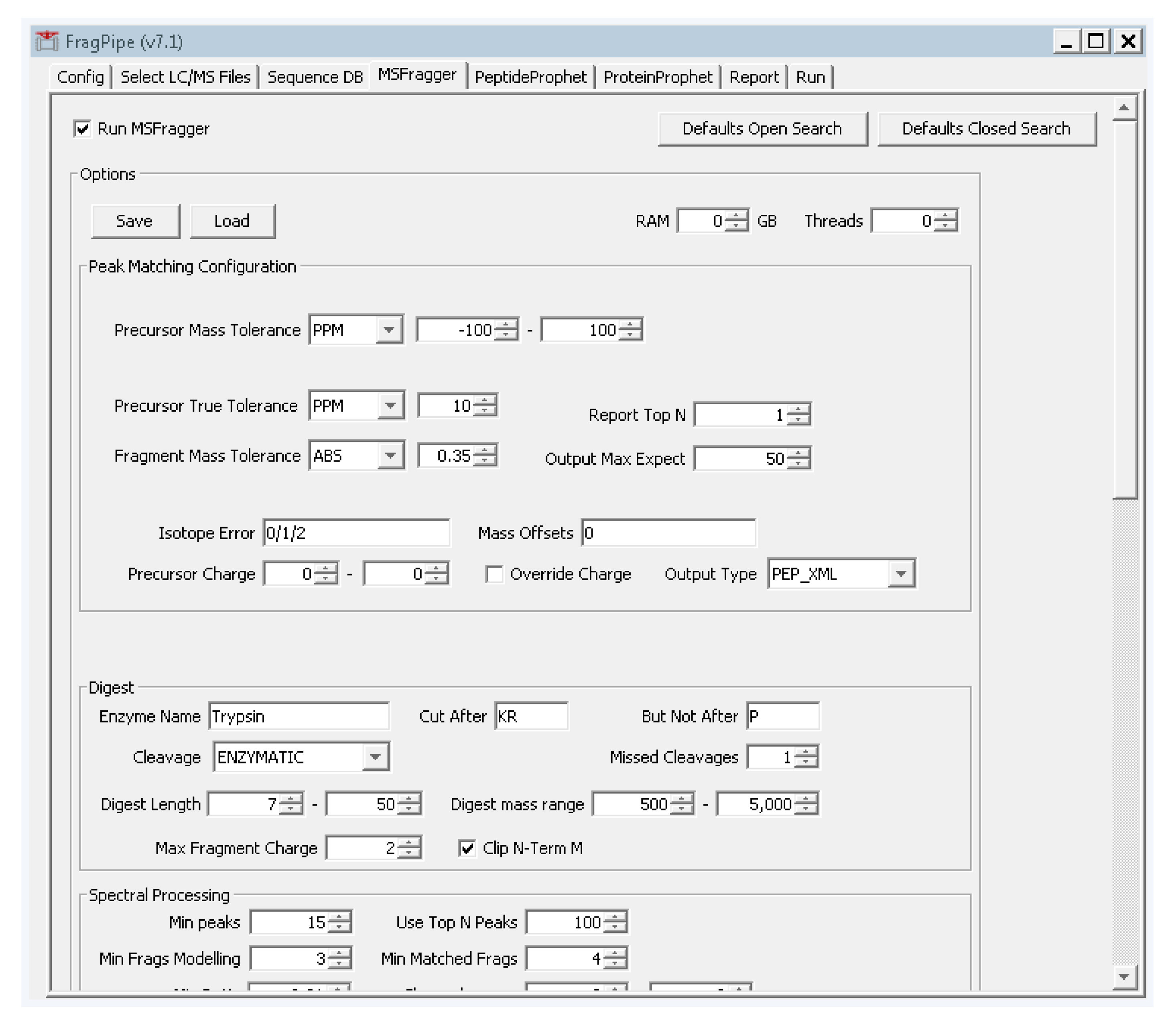
- Select the fourth tab “MSFragger”, and click the button on the top left “defaults closed search”. Two boxes will pop up asking to confirm. Click “yes” on both boxes.
- Change the precursor and fragment mass tolerances to values that reflect your instrument performance. For the tutorial data, the precursor tolerance we will use is 10 ppm. Fragmentation spectra were collected at low resolution in the ion trap, so from the dropdown box to the right of “fragment mass tolerance”, set the value to “ABS” and enter 0.35 (Figure A4). These settings are specific to the type of data collection used for the tutorial data and should be adjusted according to the expected accuracy of the data. For TripleTOF (Q-TOF, AB Sciex) data, suitable settings are 30 ppm precursor mass tolerance and 40 ppm fragment mass tolerance.
- At the top-right of the “options” section, leave the RAM and threads set to 0, and the program will determine these settings for you. You can set these parameters to reflect your computer’s available resources, but this is not required.
- Leave the remaining tabs with default settings and select the last tab “run”. Set your output file location by clicking the “browse” button, and then click “run” to start the database searches, PeptideProphet, and ProteinProphet analysis. This step will take around 1 h depending on the speed of your computer.
- Combine the PeptideProphet output files into a single result file using iProphet. In the command prompt from Section 3.2.2, type:philosopher_windows_amd64.exe iprophet data/*.pep.xml.This step will take approximately 1 h depending on the speed of your computer.
3.3. Quantify Peptides with Skyline; Time for Completion: 2 Hours
- Open Skyline by clicking the windows start button, typing “Skyline”, selecting Skyline, and hitting enter.
- On the Startup page, click the option in the top middle, “Import DDA Peptide Search”.
- Skyline will prompt you to save the document. Save the document, and then Skyline will prompt you with the “Import Peptide Search” box. Set the cutoff score to 0.99, and then click “Add Files …” and navigate to your MSFragger output folder. Select the iproph.pep.xml file. Click “Next” and Skyline will start reading the files and building your spectral library.
- Skyline will then prompt you to extract chromatograms and should find your .mzXML files. If not, browse to add them (Figure A6 in Appendix B).
- Skyline will prompt you to optionally remove any common prefix from the file names. Click “remove”, and then it will prompt you to add modifications it found in your database search results. Select the modifications you expect and want to use for quantification, in our case N-terminal acetylation, and click “next”.
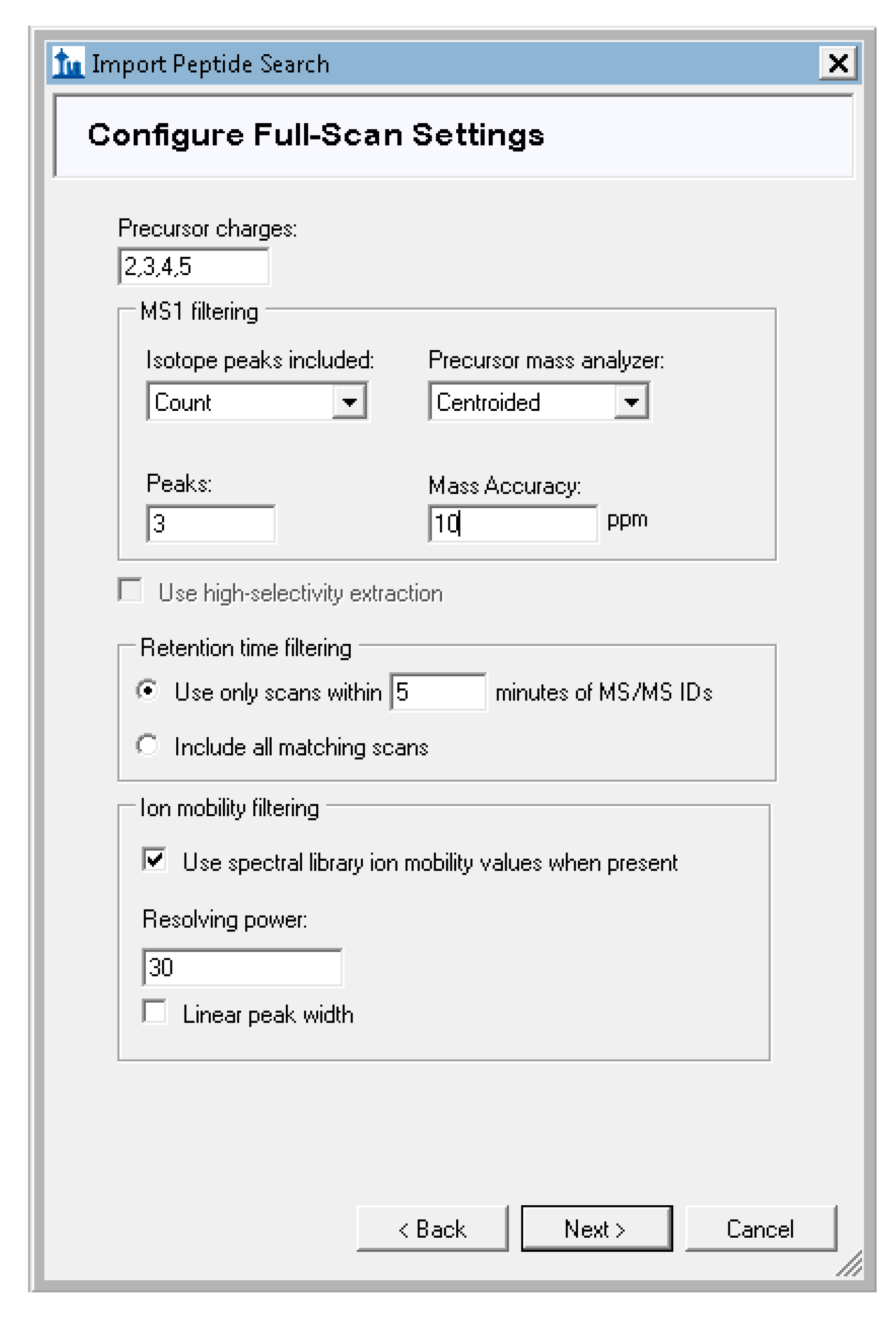
- Skyline will prompt you to configure the full-scan settings used for signal extraction. For our tutorial data, set the precursor charges to “2,3,4,5”, and leave the other defaults unchanged (Figure A7). The mass tolerance default of 10ppm here is specific to the type of data collection used for the tutorial data and should be adjusted according to the expected accuracy of the data. For TripleTOF (Q-TOF, AB Sciex) data, change this value to 30 ppm precursor mass tolerance or a value that matches the accuracy of your instrument.
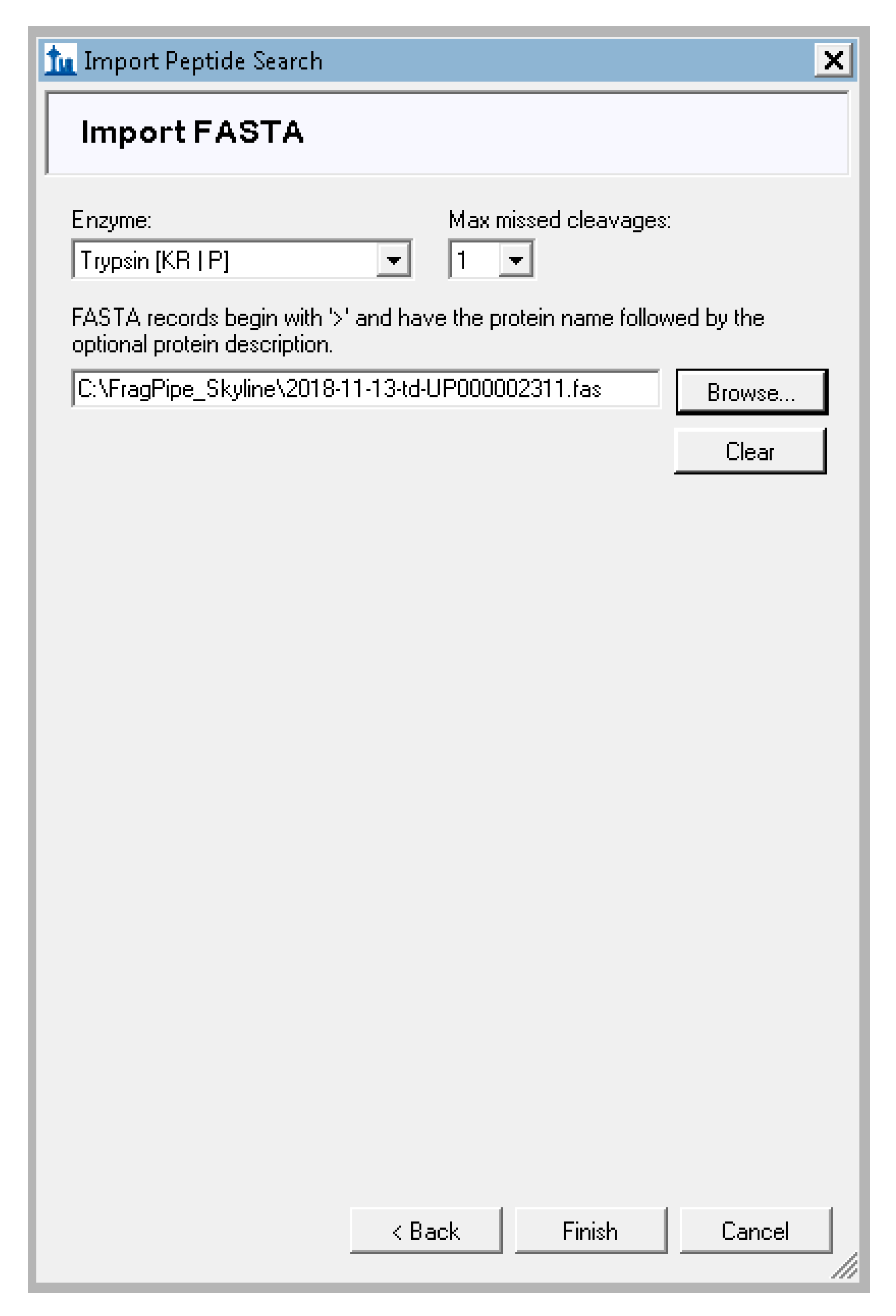
- Skyline will prompt you for the database used to search for peptides, the enzyme used to digest to proteins, and the number of missed cleavages allowed. Leave the Enzyme as “trypsin”, the missed cleavages as 1 or the value that matches your MS-Fragger search settings, and click “browse” to navigate to the FASTA file created in step 3 (Figure A8). If another protease was used to digest proteins before the mass spectrometry analysis, such as LysC, this can be specified instead of trypsin here. Click “finish” and Skyline will begin adding the proteins that match the identified peptides.
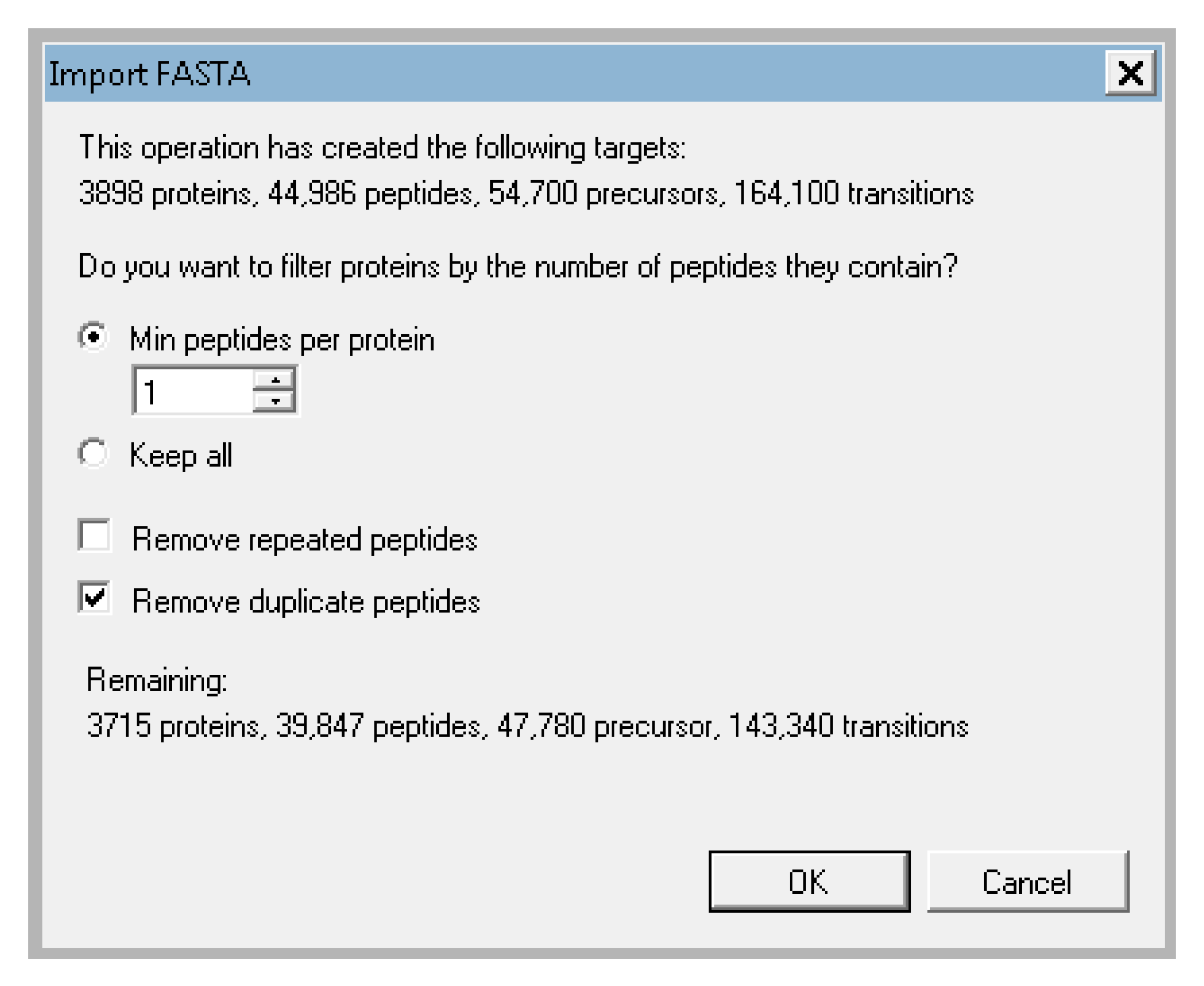
- Skyline will then prompt you about what proteins you want to keep. You can filter based on the number of proteins identified and whether or not you will allow duplicate peptides. For the tutorial data, keep the default of 1 peptide per protein, and check the box next to “remove duplicate peptides” (Figure A9). This will remove any peptide in the document that matches to multiple proteins. This is important for quantification because if the peptide could come from many proteins, we cannot be sure which protein is the true source and including such ambiguous matches in a protein’s quantification could be misleading. Skyline will then begin extracting the precursor peaks for the identified peptides. You can proceed with the next steps while Skyline continues to import the raw data. The raw data import will take about 1 h depending on the speed of your computer.
3.4. Statistical Testing with MSstats: Time for Completion: 1 Hour
- Install MSstats within Skyline by going to the “tools” menu > “tool Store”, and then selecting MSstats from the list along the left side and clicking “install”. The installer will also install R, and may take a few minutes.
- Go to the “settings” menu > “document settings”. Check the boxes next to “condition” and “BioReplicate” and click OK.
- Click on the “view” menu and select “document grid”. In the document grid popup box, click on the “views” dropdown menu and select “replicates”. Under the “condition” column, select “disease” for the PIM1 replicates, and “healthy” for the WT replicates. Under the BioReplicate column, assign the number of each biological replicate to each sample (Figure A10). Close the Document Grid box.
- Once the data has finished importing, go to the menu “tools” > “MS stats” > “group comparisons”. Skyline will take a moment to write a report file for input to MSstats. In the popup box “MSstats group comparison”, name the comparison and leave the other settings as default, then click OK (Figure A11). The “immediate window” will pop up and display the status of the process.
- After the “immediate window” displays “finished”, the MSstats output will appear in the same directory as the Skyline file.
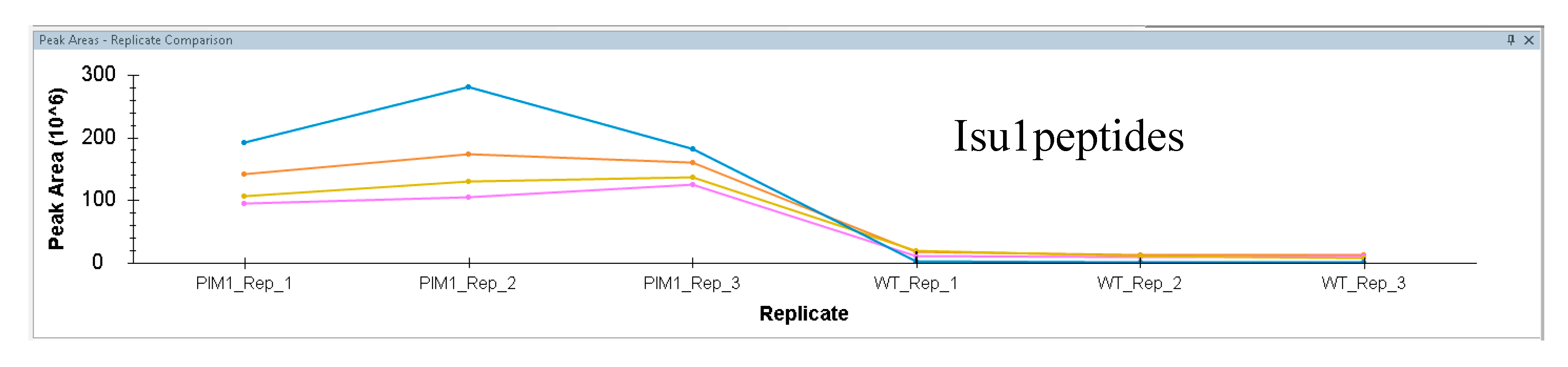
- Skyline can be used to directly inspect the changes reported by MSstats. To arrange your Skyline workspace for easy data inspection, go to the “view” menu > “arrange graphs” > “tiled”. Also add the peak area comparison window by selecting “view” > “peak areas” > “replicate comparison”. Drag your “peak areas—replicate comparison” window to the bottom of the master Skyline window and drop it over the down arrow that appears to anchor it at the bottom. Your workspace will then appear as shown in Figure 3.
4. Expected Results
Supplementary Materials
Funding
Conflicts of Interest
Appendix A




Appendix B







References
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The One Hour Yeast Proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Hebert, A.S.; Ulbrich, A.; Bailey, D.J.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. One-hour proteome analysis in yeast. Nat. Protoc. 2015, 10, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteom. 2010, 73, 2092–2123. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.G.; Mukkamalla, S.; Steen, H.; Nesvizhskii, A.I.; Gibson, B.W.; Schilling, B. PIQED: Automated identification and quantification of protein modifications from DIA-MS data. Nat. Meth. 2017, 14, 646–647. [Google Scholar] [CrossRef]
- Ong, S.-E.; Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 2007, 1, 2650–2660. [Google Scholar] [CrossRef]
- Li, Z.; Adams, R.M.; Chourey, K.; Hurst, G.B.; Hettich, R.L.; Pan, C. Systematic Comparison of Label-Free, Metabolic Labeling, and Isobaric Chemical Labeling for Quantitative Proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Hebert, A.S.; Merrill, A.E.; Bailey, D.J.; Still, A.J.; Westphall, M.S.; Strieter, E.R.; Pagliarini, D.J.; Coon, J.J. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [PubMed]
- Teo, G.; Kim, S.; Tsou, C.-C.; Collins, B.; Gingras, A.-C.; Nesvizhskii, A.I.; Choi, H. mapDIA: Preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J. Proteom. 2015, 129, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Choi, M.; Chang, C.-Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef]
- Veling, M.T.; Reidenbach, A.G.; Freiberger, E.C.; Kwiecien, N.W.; Hutchins, P.D.; Drahnak, M.J.; Jochem, A.; Ulbrich, A.; Rush, M.J.P.; Russell, J.D.; et al. Multi-omic Mitoprotease Profiling Defines a Role for Oct1p in Coenzyme Q Production. Mol. Cell 2017, 68, 970–977. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, J.G. Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods Protoc. 2019, 2, 8. https://doi.org/10.3390/mps2010008
Meyer JG. Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods and Protocols. 2019; 2(1):8. https://doi.org/10.3390/mps2010008
Chicago/Turabian StyleMeyer, Jesse G. 2019. "Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools" Methods and Protocols 2, no. 1: 8. https://doi.org/10.3390/mps2010008
APA StyleMeyer, J. G. (2019). Fast Proteome Identification and Quantification from Data-Dependent Acquisition–Tandem Mass Spectrometry (DDA MS/MS) Using Free Software Tools. Methods and Protocols, 2(1), 8. https://doi.org/10.3390/mps2010008