Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries

 ,
, 
Abstract
1. Introduction
- By introducing randomly selected nucleotides as a cost-efficient method during the construction of the synthetic library, stop codons can occur that significantly decrease its quality by lowering the number of clones expressing a full-length protein.
- The stronger the binders, the harder it is to elute them from their antigen and, hence, the best binders can be easily lost during the selection process.
- A significant bottleneck of a phage display selection is the production of sufficient amounts of bioactive monoclonal binders since the low expression level of properly folded proteins from the periplasmic space can be challenging.
2. Experimental Design
2.1. Materials
- MICROLON® microplate, 96-well, F-bottom, high-binding (Greiner, Frickenhausen, Germany; Cat. no.: 655 061)
- Cellstar®, 96 Well Suspension Culture Plate, U-bottom (Greiner, Frickenhausen, Germany; Cat. no.:650001)
- PCR grade water (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: AM9932)
- dNTP mix (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: R0191)
- Platinum™ II Hot-Start Green PCR Master Mix (2X) (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: 1400101)
- T4 DNA ligase (5 U/µL; Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: EL0011)
- Agarose (Carl Roth, Karlsruhe, Germany; Cat. no.: 11388983001)
- 100 base pair(s) (bp) ladder (New England Biolabs, Frankfurt am Main, Germany; Cat. no.: N0551G)
- NcoI Restriction Enzyme (New England Biolabs, Frankfurt am Main, Germany; Cat. no.: R0193)
- NotI Restriction Enzyme (New England Biolabs, Frankfurt am Main, Germany; Cat. no.: R0189)
- Anti-human IgG Peroxidase conjugate (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: A0545)
- Anti-M13 pIII monoclonal antibody (New England Biolabs, Frankfurt am Main, Germany; Cat. no.: E8033)
- Anti-M13 pVIII filamentous phages antibody (PROGEN, Heidelberg, Germany; Cat. no: 65197)
- Anti-mouse IgG Peroxidase conjugate (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: A8924)
- Anti-Penta-His TM monoclonal antibody (Merck Millipore, Darmstadt, Germany; Cat. no.: 70796)
- GeneRuler™ DNA Ladder Mix (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: SM0331)
- Lysozyme (Carl Roth, Karlsruhe, Germany; Cat. no.: 8259.1)
- Maximo Taq DNA Polymerase (GeneOn, Ludwigshafen, Germany; Cat. no.: S101)
- Ampicillin (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: A9393)
- Kanamycin (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: 60615)
- Trypsin (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: T1426)
- E. coli TG1: F′ [traD36 proAB+ lacIq lacZΔM15] supE thi-1 Δ(lac-proAB) Δ(mcrB-hsdSM)5, (rK-mK-) (Lucigen, Middleton, USA; Cat. no.: 60502).
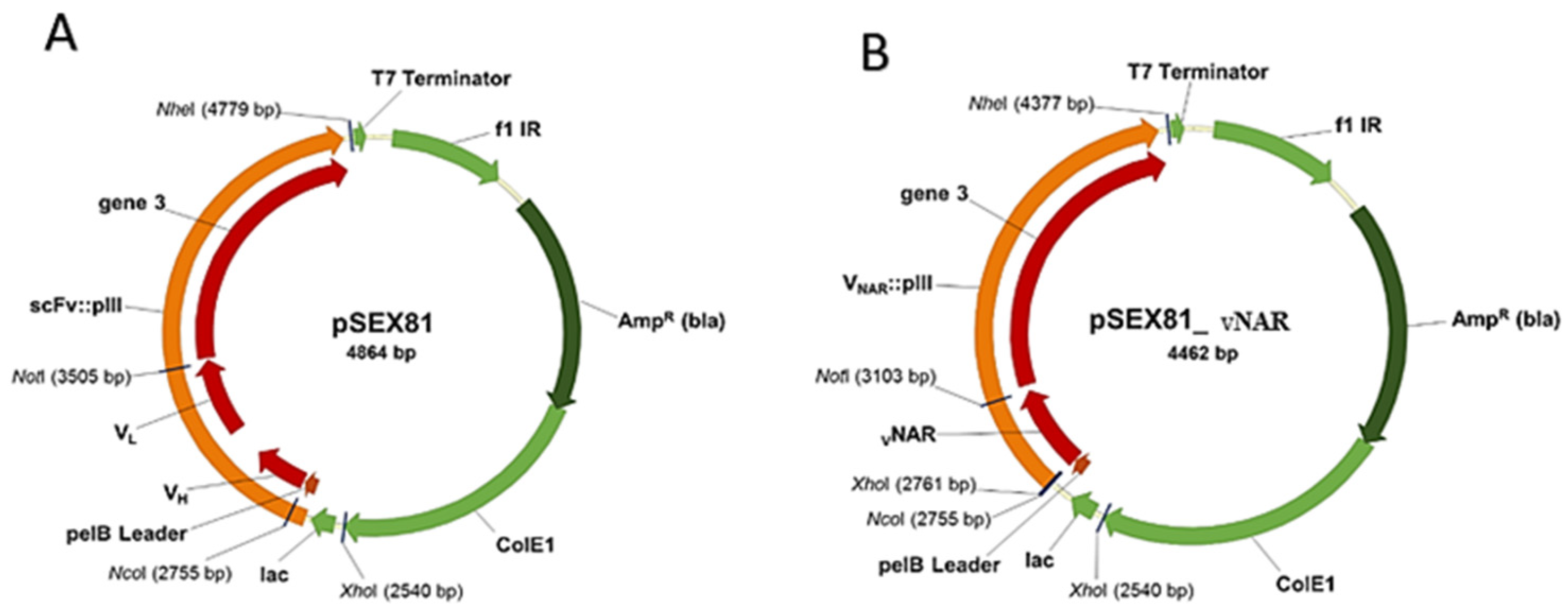
- Phagemid pSEX81 (PROGEN, Heidelberg, Germany; Cat. no: PR3005)
- Hyperphage (PROGEN, Heidelberg, Germany; Cat. no. PRHYPE-XS)
- Oligonucleotide primers for PCR are listed in Table 1.
- Gene Pulser®/MicroPulser™ Electroporation Cuvettes, 0.2 cm gap (Micropulser™, Bio-Rad, Munich, Germany; Cat. no.: 1652082)
- PEG-8000 (Sigma-Aldrich Chemie GmbH, Munich, Germany; Cat. no.: 729108-1G)
- Monarch™ DNA Gel Extraction Kit (New England Biolabs, Frankfurt am Main, Germany; Cat. no.: T1020G)
- M13KO7 helper phages (New England BioLabs, Frankfurt am Main, Germany; Cat. no.: N0315S)
- 1-Step™ Ultra 3,3′,5,5′-Tetramethylbenzidine-enzyme-linked immunosorbent assay (TMB-ELISA) (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: 34028)
- 6X DNA Gel Loading Dye (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: R0611)
- Agar-Agar Carl Roth (Carl Roth, Karlsruhe, Germany; Cat. no.: 6494.4)
- Agarose (Biozym Scientific, Oldendorf, Germany; Cat. no.: 840004)
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Carl Roth, Karlsruhe, Germany; Cat. no.: 9105.4)
- Roti-Safe Gel Stain (Carl Roth, Karlsruhe, Germany; Cat. no.: 3865.1)
- Tryptone/Peptone (Carl Roth, Karlsruhe, Germany; Cat. no.: 8952.4)
- Tween® 20 (Carl Roth, Karlsruhe, Germany; Cat. no.: 9127.2)
- Yeast extract (Carl Roth, Karlsruhe, Germany; Cat. no.: 2363.1)
- Roti®-Prep Plasmid MINI (Carl Roth, Karlsruhe, Germany; Cat. no.: HP29.2)
- GeneJet PCR purification kit (Thermo Fisher Scientific, Darmstadt, Germany; Cat. no.: K0702)
2.2. Oligonucleotides
2.3. Equipment
- Toptable Centrifuge 5427R (Eppendorf, Hamburg, Germany; Cat. no.: 5409000012)
- Electrophoresis gel tank and power pack (VWR Peqlab, Lutterworth, UK; Cat. no.: 700-0444)
- Electroporator Gene Pulser® (Micropulser™) (Bio-Rad, Munich, Germany; Cat. no.: 1652100)
- Fusion FX Gel Documentation (Vilber Lourmat, Collégien, France; Cat.: no.: 826)
- New Brunswick Innova 4200 Incubator (Eppendorf, Hamburg, Germany; Cat. no.: NB-4200)
- NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA; Cat.: 25627)
- PCR Cycler Eppendorf Mastercycler (Eppendorf, Hamburg, Germany; Cat. no.: 5332)
- SpectraMax M Series Multi-Mode Microplate Readers (Molecular Devices, San Jose, CA, USA; Cat. no.: MLDVM2)
2.4. Software
- Vector NTI Advance 11
- GraphPad Prism version 7 Software (GraphPad Software Inc., La Jolla, CA, USA)
- CLC Main Workbench 8.0.1 (QIAGEN Redwood City, CA, USA)
3. Procedure
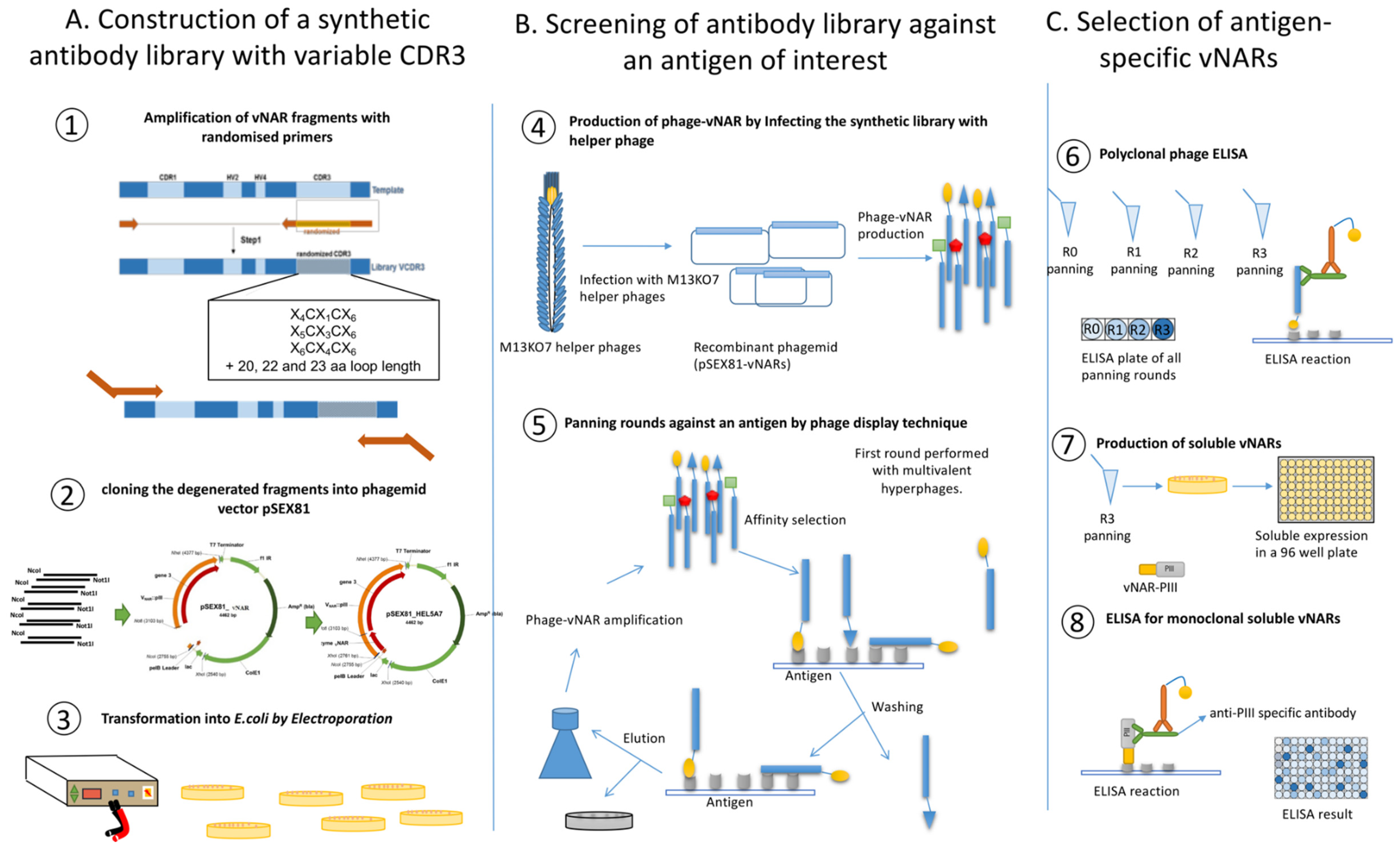
3.1. In vitro Synthesis of Variable DNA Fragments and Cloning into the Phagemid pSEX81
- For each reaction with different randomized reverse primer, add the followings material to nuclease-free water to make a final volume of 50 µL in a PCR tube on ice: 25 µL of Platinum™ II Hot-Start Green PCR Master Mix, 10 µL Platinum™ GC Enhancer (provided by PCR master mix), 5 ng of template DNA (HEL-5A7) and finally add 1 µL of each 10 µM primers (final concentration 0.2 µM). Use ForFR1_NcoI as forward primer and one of the randomized reverse primers (RevRan_12 to RevRan_23) for each reaction. Mix well and briefly centrifuge the contents.
- Perform PCR with the following set up: initial denaturation at 94 °C for 2 min. Then proceed for 20 cycles as follows: 5 s at 98 °C, annealing at 60 °C for 15 s and elongation of fragments for 15 s at 68 °C.
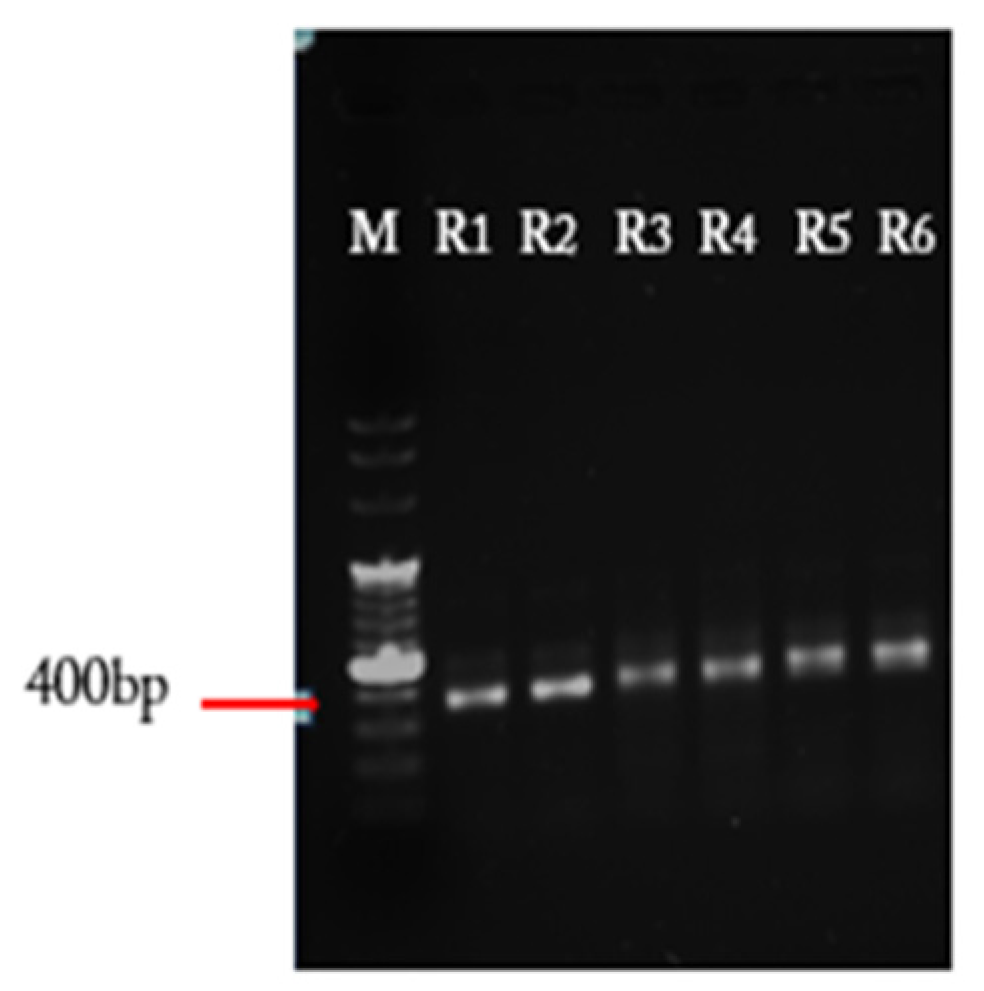
- Load PCR samples onto a 1% agarose gel and run the gel at 110 V. Inspect the gel under UV light. Excise the bands around 350 bp length and extract them from the agarose gel using the DNA gel extraction kit, according to manufacturer’s protocol. Elute the DNA of each PCR reaction with 10 µL nuclease-free water.
- Use the eluted PCR product for a second PCR to introduce restriction digestion sites. Proceed as it is described in step 2, use ForFR1_NcoI and RevFR4_NotI as primers for the reaction.
- Digest both, the PCR products and the pSEX81 plasmid with NcoI and NotI. For this, prepare a total volume of 50 µL digestion reaction for the PCR products (at least 4 µg) by adding 2 µL (20 units) of each enzyme NcoI and NotI, 5 µL of 10X NEB3 buffer and adjust the volume reaction up to 50 µL with nuclease-free water. For the plasmid, digest 20 µg of the pSEX81 vector, add 15 µL of 10X NEB3 buffer, 10 µL (100 units) of each enzyme NcoI and NotI and add nuclease-free water up to 150 µL. Incubate for 3 h at 37 °C following 10 min at 65 °C.
- After restriction digestion, purify DNA fragments using GeneJet PCR purification kit according to the manufacturer’s instruction. Elute each purified digested sample in 20 µL water.
- Run the digested plasmid on a 1% agarose gel and cut out the target band quickly with a scalpel on a UV-transilluminator table. Purify the DNA fragments from the agarose gel using the Monarch™ DNA Gel Extraction Kit, following the manufacturer’s instructions. Elute in 100 µL preheated sterile H2O. Determine the concentration by Nanodrop 1000 spectrophotometer.

- PAUSE STEP: The extracted and purified digested DNA fragments can be stored at −20 °C until use.
- Ligate the digested fragments by preparing following material: Calculate the amount of needed insert for 100 ng of the plasmid with these molar ratios: 1:0.5, 1:1, 1:2 vector: insert. Add 1 µL of ligase, 1 µL of Ligase buffer (10X) and water to 10 µL. Keep at 16 °C overnight following 10 min at 65 °C.
- Transform 2 µL of ligation reaction by electroporation into TG1 cells. Plate 100 µL of 10−2, 10−3, and 10−4 dilutions on 2X TY-GA agar plates and incubate overnight at 37 °C.
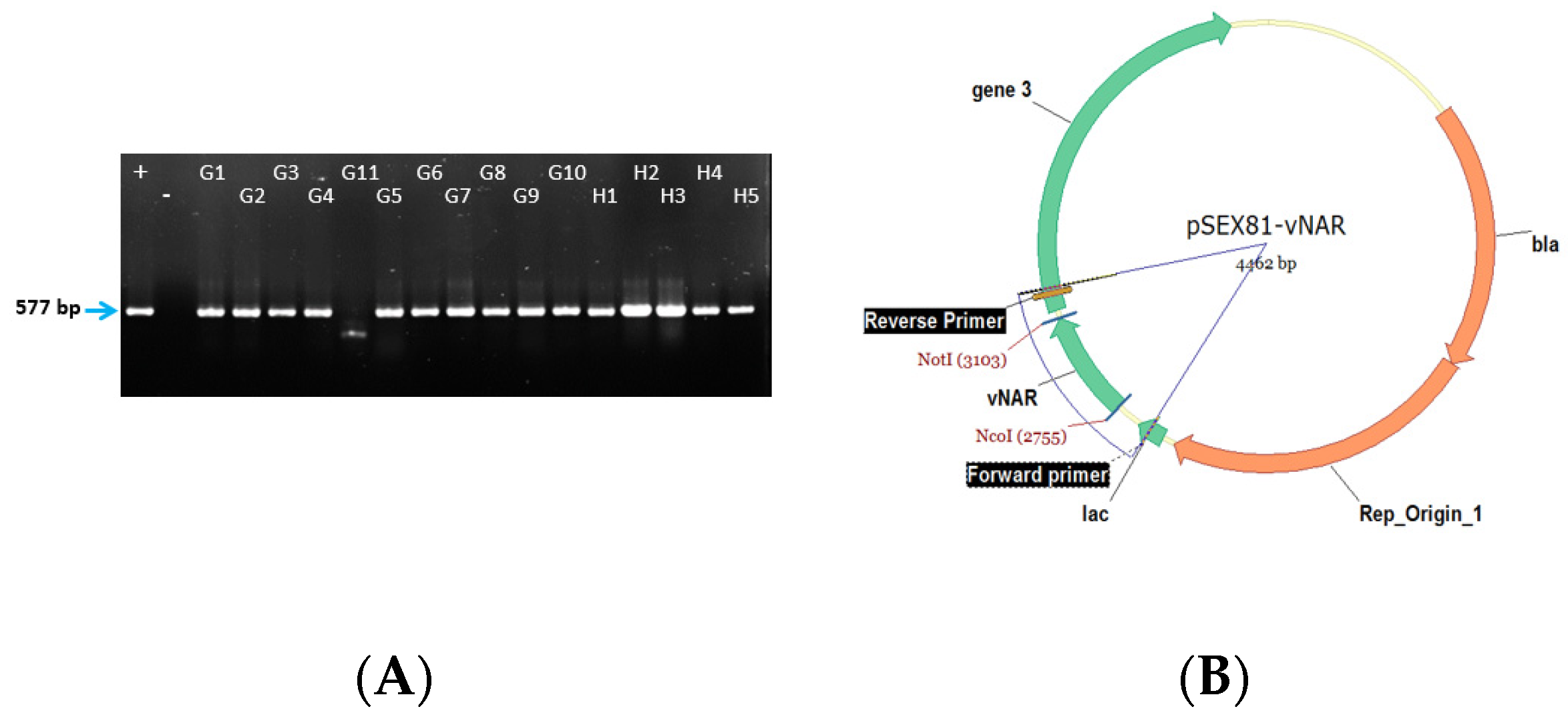
- Measure the cloning efficiency by colony PCR with For_pSEX and Rev_pSEX primers. For this, pick 50 individual clones, mix each clone with 20 µL of water and cook them at 95 °C for 10 min. Later centrifuge them and take 2 µL of the supernatant as a template for the PCR.
- Based on the result of cloning efficiency, prepare the final ligation reaction for the best-determined ratio. For each insert with specific CDR3 length, add 2 µg of plasmid and an appropriate amount of each insert into 100 µL reaction volume. Incubate at 16 °C for 16 h, heat for 10 min at 65 °C and purify using GeneJet PCR purification kit. Elute each reaction in 20 µL and pool all the sample together.

- PAUSE STEP: After stopping the reaction, the mix can be stored at −20° C until further use.

3.2. Transformation in Electrocompetent TG1 and Library Construction.

- CRITICAL STEP: In order to increase the transformation efficiency, prepare electrocompetent cells on the same day of transformation and avoid freezing the cells before transformation.

3.2.1. Preparation of Electrocompetent TG1.

- CRITICAL STEP: Chill all the material including HEPES, glycerol, H2O, pipettes and tips at 0 °C. Centrifuge and rotor must be precooled to 2 °C.
- 12.
- Pick a fresh clone of E. coli TG1 from 2X TY-agar plate into 20 mL 2X TY medium in a 125 mL flask and incubate at 37 °C with vigorous shaking at 220 rounds per min (rpm) overnight.
- 13.
- The next day, add the overnight culture to 1 L fresh 2X TY in a 5 L flask (with starting OD600 at around 0.1) and incubate at 37 °C and 180 rpm until an optical density OD600 of 0.7.
- 14.
- Distribute the flask content equally into two sterile 500 mL centrifuge bottles and cool down on ice for 30 min.
- 15.
- Centrifuge for 5 min with 5000× g at 2 °C. All subsequent centrifugation steps should be done with 5000× g at 2 °C.
- 16.
- Remove the supernatant of both bottles carefully and re-suspend each pellet in 500 mL cold H2O/HEPES (see reagent setup). Add a sterile magnetic stir bar to each bottle to completely dissolve the pellets on a magnetic stirrer.
- 17.
- Centrifuge for 10 min and repeat the previous step with another 500 mL cold H2O/HEPES.
- 18.
- Discard the supernatant and resuspend each pellet in 50 mL cold glycerol/HEPES. Pool the bacteria suspension of both bottles into a fresh sterile centrifugation bottle without transferring the stir bars.
- 19.
- Centrifuge for 15 min. Remove the supernatant and re-suspend the pellet in 1 mL cold glycerol/HEPES (see reagent setup).
- 20.
- Distribute this volume into 300 µL aliquots. Use immediately for transformation.
3.2.2. Transformation of Electrocompetent E. coli TG1 by Electroporation

- CRITICAL STEP: Cool at least 6 cuvettes and slides of the gene pulser down to 2 °C. For each control and test sample, prepare 0.950 mL of SOC in 2 mL tubes and 5 sterile 100 mL Erlenmeyer flasks with 25 mL SOC each for the samples.
- 21.
- Apply this setting for the gene pulser: 2.5 kV and 25 µF.
- 22.
- For negative control, dry the aluminum electrodes of the first cuvette with tissue paper, add 50 µL of electrocompetent cells to the cuvette, put the cuvette in the holder, push in into the gene pulser and start the electric pulse. Now, the cells are very fragile and you need to add pre-warmed SOC medium immediately. Rinse the cuvette a few times with SOC medium and put it quickly into the incubator.
- 23.
- For the positive control add 1 µL of highly pure pSEX81 plasmid to 50 µL of the cells and perform the electroporation as described above.
- 24.
- Add 10–15 µL of purified ligation mixture to 300 µL of electrocompetent E. coli. Gently tab the bottom of cuvette on the bench to avoid bubble formation. After electroporation, immediately transfer the cells into the 125 mL flask and wash the cuvette with the prewarmed medium.
- 25.
- Repeat step 24 for the other ligation reactions.
- 26.
- Shake the entire samples at 37 °C in 220 rpm for 45–60 min.
- 27.
- Pool the library and plate 100 µL of 10−2 to 10−5 dilutions of the library, 10−1 and 10−2 dilutions of the positive control and 100 µL of the undiluted negative control on 2X TY-GA plates.
- 28.
- Centrifuge the rest of library suspension at 3220× g for 10 min and discard the supernatant. Resuspend the pellet in 5 mL SOC medium and plate on five 150-mm 2X TY-GA plates.
- 29.
- Incubate the plates overnight at 37 °C.
- 30.
- To estimate the transformation titer, plate 100 μL of 10−3 and 10−4 dilutions of the cells on 2X TY-GA agar plates.
- 31.
- The transformation titer is calculated as:
- 32.
- Centrifuge the remaining cells (2000× g, 15 min), and resuspend the pellet in 1000 μL of 2X TY medium. Plate the suspended bacteria on a 150 mm diameter 2X TY-GA agar plate and incubate overnight at 37 °C.
- 33.
- The next morning, add 5 mL of SB medium to the 150 mm diameter agar plates and scrape the bacteria using flame-sterilized glass spreader. Add glycerol to the final concentration of 10% glycerol and mix well.
- 34.
- Prepare 1 mL aliquots, and store at −80 °C.
3.3. Phage Preparation and Selection of vNARs against Antigen by Phage Display
3.3.1. Packaging of Synthetic Library Employing Hyperphage (M13KO7ΔpIII).
- 35.
- Thaw aliquot of frozen antibody library on ice. Add it to 500 mL 2X TY-GA medium. The initial OD600 should be approximately 0.1. Grow the culture at 37 °C, 250 rpm in a 2 L glass flask until OD600 = 0.5 (approximately 1.5–2 h).
- 36.
- Add 1 × 1012 M13KO7 hyperphages to the culture and incubate at 37 °C without shaking for 15 min following 30 min vigorous shaking.
- 37.
- Spin the culture at 3200× g for 15 min and discard the supernatant.
- 38.
- Resuspend pellets in 500 mL of 2X TY-AK medium.
- 39.
- Grow for 16–20 h at 28 °C, 250 rpm in a 2 L glass flask.
- 40.
- The next day, centrifuge the culture for 20 min at 5500× g and 4 °C.
- 41.
- Add 1/4 volume of PEG/NaCl solution (see Reagent setup) to precipitate the produced phage binder from the culture supernatant.
- 42.
- Following four hours of incubation on ice, pellet the antibody phages by centrifugation for 60 min at 5500× g and 4 °C.
- 43.
- Re-suspend the white phage pellet in 1 mL PBS.
- 44.
- To remove E. coli particles and bacterial debris, centrifuge the solution three times for 5 min.
- 45.
- Collect the supernatant and transfer it to a new 1.5 mL tube containing 250 µL of PEG/NaCl.
- 46.
- Incubate it on ice for 30 min. Then pellet the precipitated phages by centrifugation at 17,900× g for 10 min and discard the supernatant.
- 47.
- To remove the final traces of PEG, centrifuge again for another 2 min at 17,900× g and carefully remove the remaining supernatant.
- 48.
- Re-suspend the phages in 0.5 mL PBS. This supernatant contains the phage particles. Store them at 4 °C. These phages are used for the first round of panning.
3.3.2. Panning of vNAR Library against an Antigen (Here TNF Alpha Was Used)
- 49.
- For the first panning round, use 4 µg protein/well per panning, for the following rounds use 1 µg protein/well for more stringent conditions. Dissolve the antigen and incubate in a microtiter plate well overnight at 4 °C. Coating conditions (incubation temperature and coating buffer) should be set-up for the antigen of interest. Prepare overnight culture of TG1 cells grown on minimal medium (M9) by transferring 1 colony from fresh culture to 5 mL LB. Growth of TG1 on minimal medium selects for cells with F’ factor.
- 50.
- For biopanning of the synthetic library, coat 2 µg of the antigen TNF alpha in 150 µL of 50 mM NaHCO3 (pH 9.6) on a 96-well well plate incubated at 4 °C overnight
- 51.
- The following day, remove the coating solution from the plate by tapping on a clean tissue. Wash the coated well three times with PBST in an ELISA washer or squirt bottle and block with 300 µL MPBST for 2 h at room temperature (RT).
- 52.
- After blocking, wash the well once with PBST and one to three times with PBS.
- 53.
- For binding of the antibody phages, incubate 1011 antibody phages in 200 µL blocking buffer for 1 h without shaking and 1 h at RT and gentle agitation (200 rpm). Meanwhile, inoculate 5 mL 2X TY with 500 µL of the overnight culture of E. coli TG1 and grow at 37 °C, 250 rpm to an OD600 0.5. The culture can be kept at 4 °C until use. For the following round of panning change the blocking buffer to BSA-PBST to eliminate nonspecific binding of phages to the blocking buffer.
- 54.
- Discard the unbound phages by washing 10 times with PBST and 10 times with PBS for 2 min each. Increase the Tween® 20 concentration by 0.1% for each washing step. For the subsequent panning rounds, increase the washing steps to 15 (round 2) and 20 (round 3 and 4) steps with PBST, respectively.
- 55.
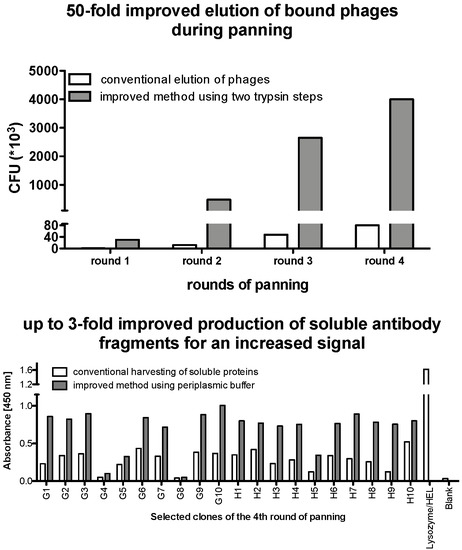
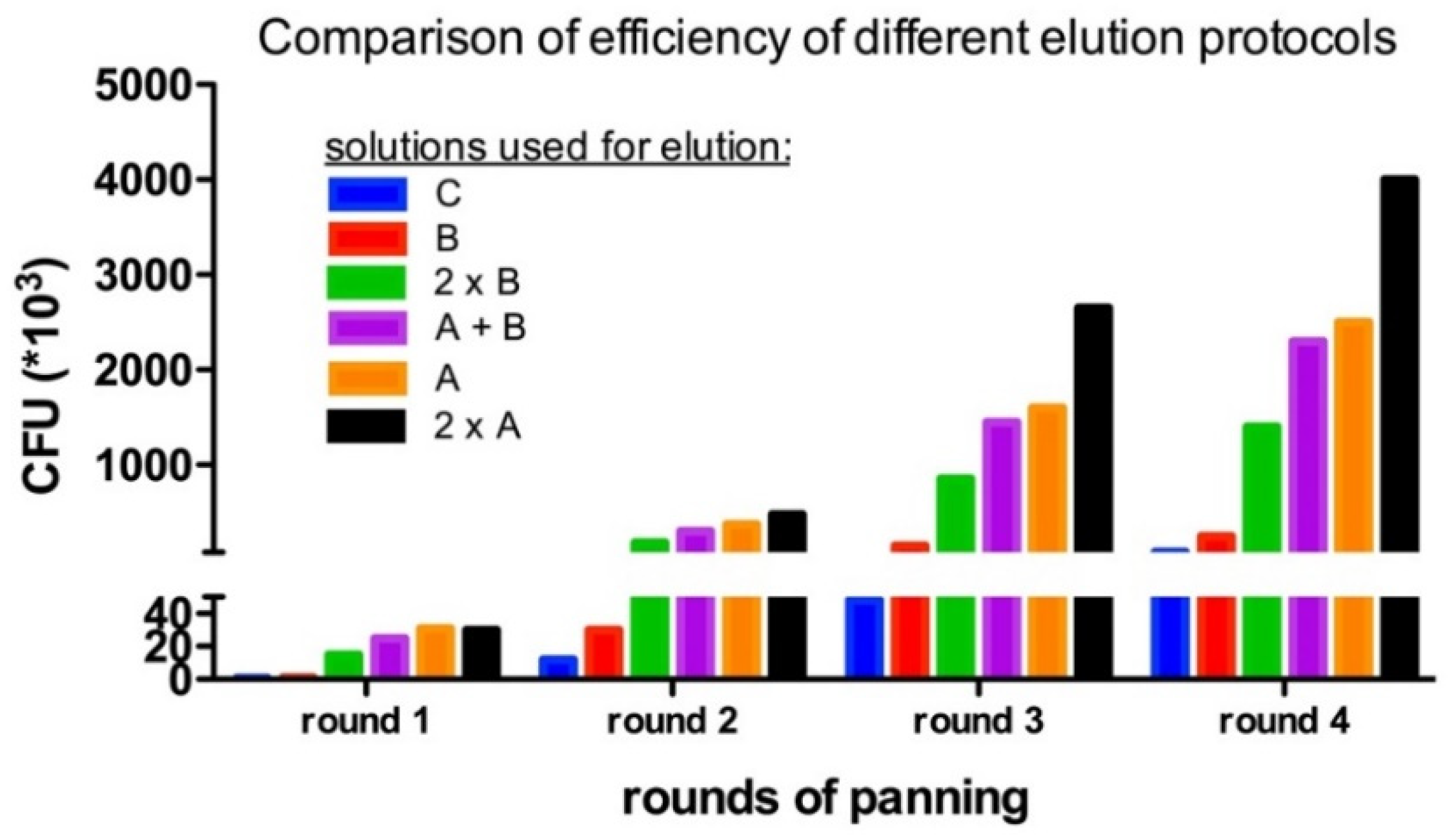
- Elute phage particles by using 50 µL trypsin solution (corresponding to Solution A in Table 2) for 30 min at 37 °C. Repeat this step two times. Decrease the incubation time to 20 min. This will ensure the detachment of all bound phage particle and increase the yield of the panning process. Remove the supernatant containing the eluted phages and store it at 4 °C for the infection.
- 56.
- Add 100 µL of a log-phased E. coli TG1 to the trypsinated well and infect 500 µL of a log-phased E. coli TG1 culture in 2X TY medium with the eluted phages from step 55. Incubate both, first for 30 min at 37 °C without shaking and then the following 30 min with moderate shaking (250 rpm). Combine both suspensions.
- 57.
- After infection, plate 100 µL of the total volume from step 56 in serial dilutions (10−2 to 10−4 in 2X TY medium) on 2X TY-GA agar plates and incubated overnight at 37 °C.
- 58.
- Centrifuge the rest of the suspension for 20 min at 3200× g at RT.
- 59.
- Suspend the pellet in 1000 µL 2X TY-GA medium and incubate overnight at 30 °C and 200 rpm.
- 60.
- The next day, centrifuge the suspension for 10 min at 3200× g and suspend the pellet in 1000 µL fresh 2X TY-GA medium.
- 61.
- Scrap the colonies from the agar plates and add to the suspension. Add 100 µL glycerol (100%) and mix thoroughly.
- 62.
- Prepare 100 µL aliquots and store at –20 °C until further use.
3.3.3. Packaging of Phagemid
- 63.
- For the subsequent round of panning, the eluted phages should be packaged and reamplified. Inoculate 1 mL of 2X TY-GA medium with 20 µL of the glycerol stock obtained after panning. Grow the culture for about 3 h at 37 °C 200 rpm.
- 64.
- Infect the cells with 2 × 109 cfu (colony forming units) of M13KO7 helper phages and incubate for 1 h at 37 °C and 200 rpm.
- 65.
- To remove the medium, centrifuge the infected culture for 30 min at 3200× g and 4 °C. Discard the supernatant.
- 66.
- Suspend the pellet in 500 µL 2X TY/AK medium and incubate for 16 h at 30 °C 200 rpm for phage production.
- 67.
- The next day, pellet the bacteria twice by centrifugation for 10 min at 3200 rpm and 4 °C to precipitate the phages. Add 1/5 volume of PEG/NaCl solution to the supernatant and incubate for 1 h on ice.
- 68.
- To pellet the phages, centrifuge the solution for 1 h at 3200× g and 4 °C.
- 69.
- Resuspend the white phage pellet in 100 µL PBS and store at 4 °C.
3.3.4. Phage Titration.
- 70.
- Make serial dilutions of the phage suspension in PBS. The number of eluted phage depends on several parameters (e.g., antigen, library, panning rounds, washing stringency).
- 71.
- Infect 50 µL bacteria with 10 µL of different phage dilutions (10−6, 10−7, 10−8) and incubate 30 min at 37 °C.
- Note: Check all solutions for phage contamination. To check the PBS or PEG solutions, use 10 µL of these solutions for E. coli “infection”. In parallel, plate out non-infected TG1 to check the bacteria.
- 72.
- Plate the 60 µL infected bacteria on 2X TY-GA agar plates.
- 73.
- Incubate the plates overnight at 37 °C.
- 74.
- Count the colonies and calculate the cfu or cfu/mL titer according to the dilution:
3.4. Screening and Selection of Antigen Binders.
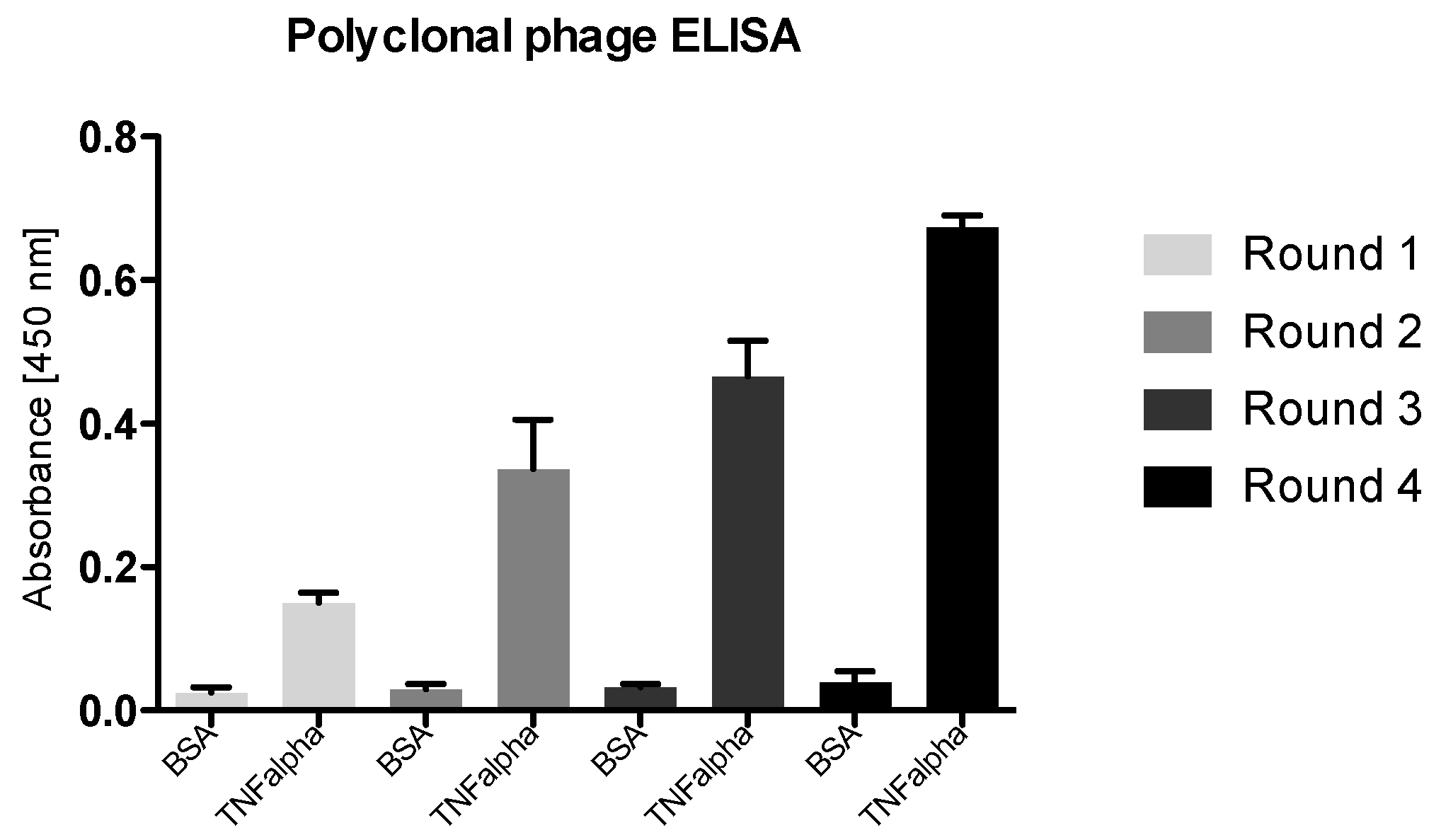
3.4.1. Polyclonal Phage ELISA
- 75.
- For this, coat a 96-well plate with 50 ng of TNF alpha in 100 µL 50 mM NaHCO3 (pH 9.6) in duplicate for each round of panning and incubated overnight at 4 °C.
- 76.
- The following day, wash the coated wells three times with 200 µL PBST and then block with 300 µL MPBST for 2 h at RT.
- 77.
- After three washing steps with each 200 µL PBST, add 109 cfu phage amplificate from each panning round to each well and incubate the plate for 1.5 h at RT.
- 78.
- After incubation, wash the wells three times with 200 µL PBST
- 79.
- Add 100 µL of a 1:100 dilution of anti-pVIII monoclonal antibody in blocking buffer and incubate for an additional 1 h at RT.
- 80.
- Wash the plates as above, and add 100 µL of a 1:10,000 dilution of the secondary antibody anti-mouse IgG peroxidase conjugate in blocking buffer and incubate for a further hour at RT.
- 81.
- After three washing steps add 100 µL of 1-Step™ Ultra TMB-ELISA substrate for color development to each well.
- 82.
- Stop the reaction by the addition of 100 µL of 1 M sulfuric acid (H2SO4).
- 83.
- Measure the absorbance at 450 nm using a SpectraMax Reader or a similar absorption reader.
3.4.2. Production of Soluble Monoclonal Antibody Fragments in Microtitre Plates
- 84.
- Take 20 µL of phage-antibody particles either from the last round of panning or the one with the highest enrichment, add 50 µL of trypsin solution and incubate for 30 min at 37 °C.
- 85.
- Infect 500 µL exponentially growing TG1 bacteria for 30 min at 37 °C.
- 86.
- Prepare different dilution series; 10−2–10−4 and plate them on 2X TY-GA and incubate overnight at 37 °C.
- 87.
- The next day, fill each well of a 96 well U-bottom polypropylene plate with a total volume of 1 mL per well with 100 µL 2X TY-GA.
- 88.
- Inoculate each well with an individual clone using sterile tips. Consider two wells for negative control to check the contamination of the wells and two wells for positive clone (containing pSEX81-Hel5A7). Seal the plate with a breathable sealing film.
- 89.
- Incubate overnight at 37 °C and 250 rpm.
- 90.
- Transfer 10 µL of overnight culture to a new 96 well plate and add 150 µL 2X TY-GA. Incubate at 37 °C and 250 rpm for approximately 2–3 h to reach an OD600 of 0.7–0.9. To the rest of the overnight culture add 10 µL glycerol, mix well, and store at −80 °C.
- 91.
- Centrifuge the plate for 10 min at 4000 rpm, remove the medium carefully and add 150 µL 2X TY medium, suspend the pellet and centrifuge again.
- 92.
- Add 150 µL 2X TY-A with 100 µM IPTG and incubate overnight at 30 °C and 250 rpm.
- 93.
- Centrifuge for 10 min at 4000 rpm. Transfer the antibody fragment containing supernatant to a new polypropylene plate (first supernatant) and store at 4 °C.
- 94.
- Add 100 µL of the periplasmic buffer to the pellet, resuspend and incubate on ice for 30 min.Centrifuge the plate, remove the supernatant (second supernatant) and combine it with the first supernatant. Keep at 4 °C until the next step.
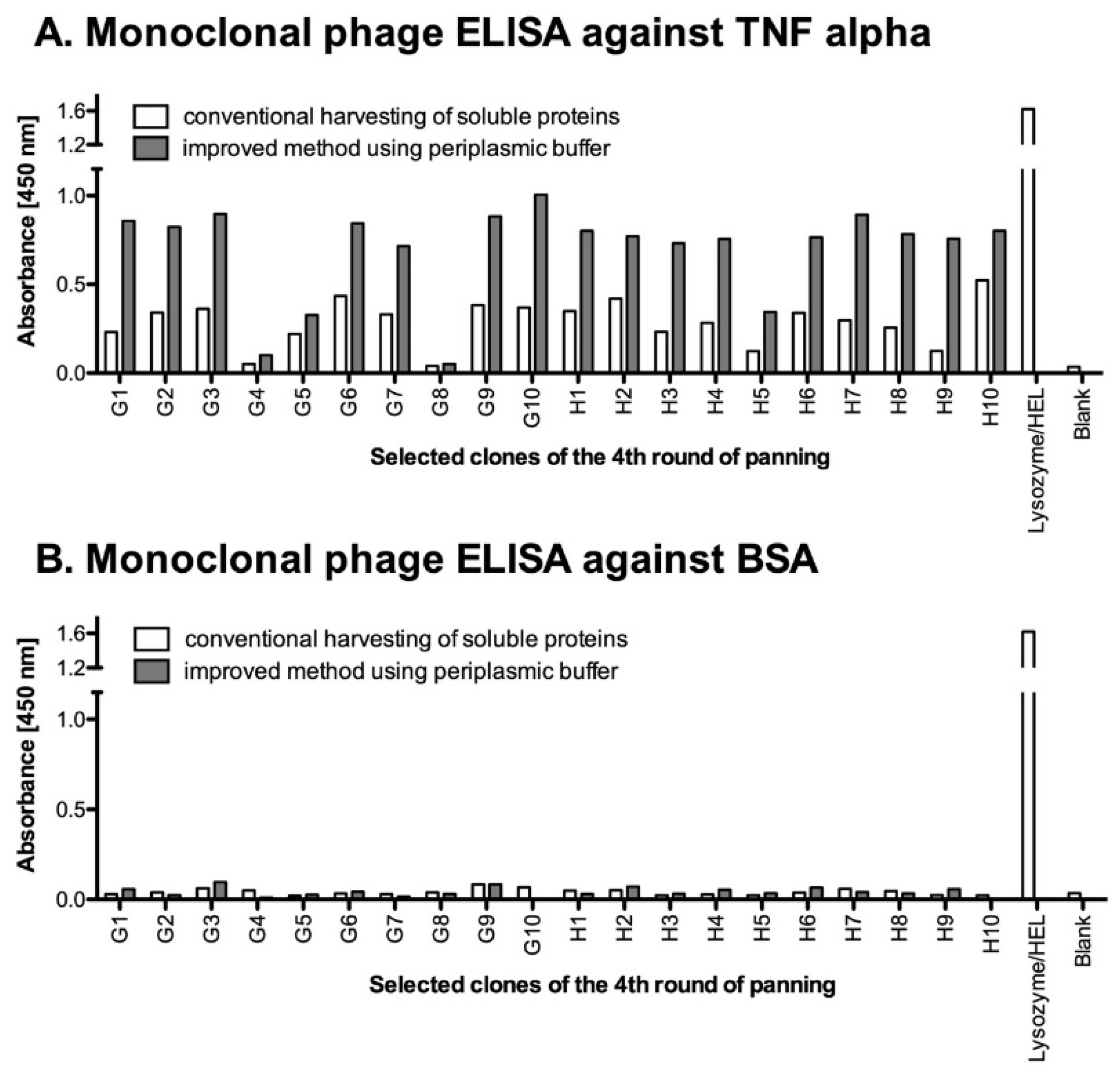
3.4.3. ELISA for Detection of Antigen Binding Monoclonal Soluble Antibody Fragments
- 95.
- Coat 50 ng of antigen into 96 well plates in 100 µL of carbonate buffer (50 mM NaHCO3; pH 9.6) or PBS for coating. For the positive control, coat 100 µL of 1 mg/mL of lysozyme and 50 ng of BSA as a negative control. Incubate overnight at 4 °C or 1 h at 37 °C.
- 96.
- Next day, wash the coated wells three times with 200 µL PBST and then block with 300 µL MPBST for at least 1 h at RT.
- 97.
- Mix 100 µL of soluble antibody fragments (combined first and second supernatant) with 100 µL of blocking buffer and add them to coated wells. Incubate for 90 min at RT.
- 98.
- Wash 3 times with PBST.
- 99.
- As a primary antibody, use a 1:1000 dilution of an anti pIII monoclonal antibody and incubate for 1 h at RT.
- 100.
- Add secondary antibody and proceed with the steps 80–83 of Section 3.4.1.
3.4.4. Sequencing
- 101.
- To determine the nucleotide sequence of plasmid DNA, DNA samples should be sent to sequencing. The sequencing of our sample was performed at GATC BIOTECH AG (Köln, Germany). The resulting data of the sequencing can be analyzed using the software of Vector NTI or CLC Main Workbench 8 or any similar software.
4. Expected Results
4.1. In Vitro Synthesis of Variable DNA Fragments
4.2. Colony PCR
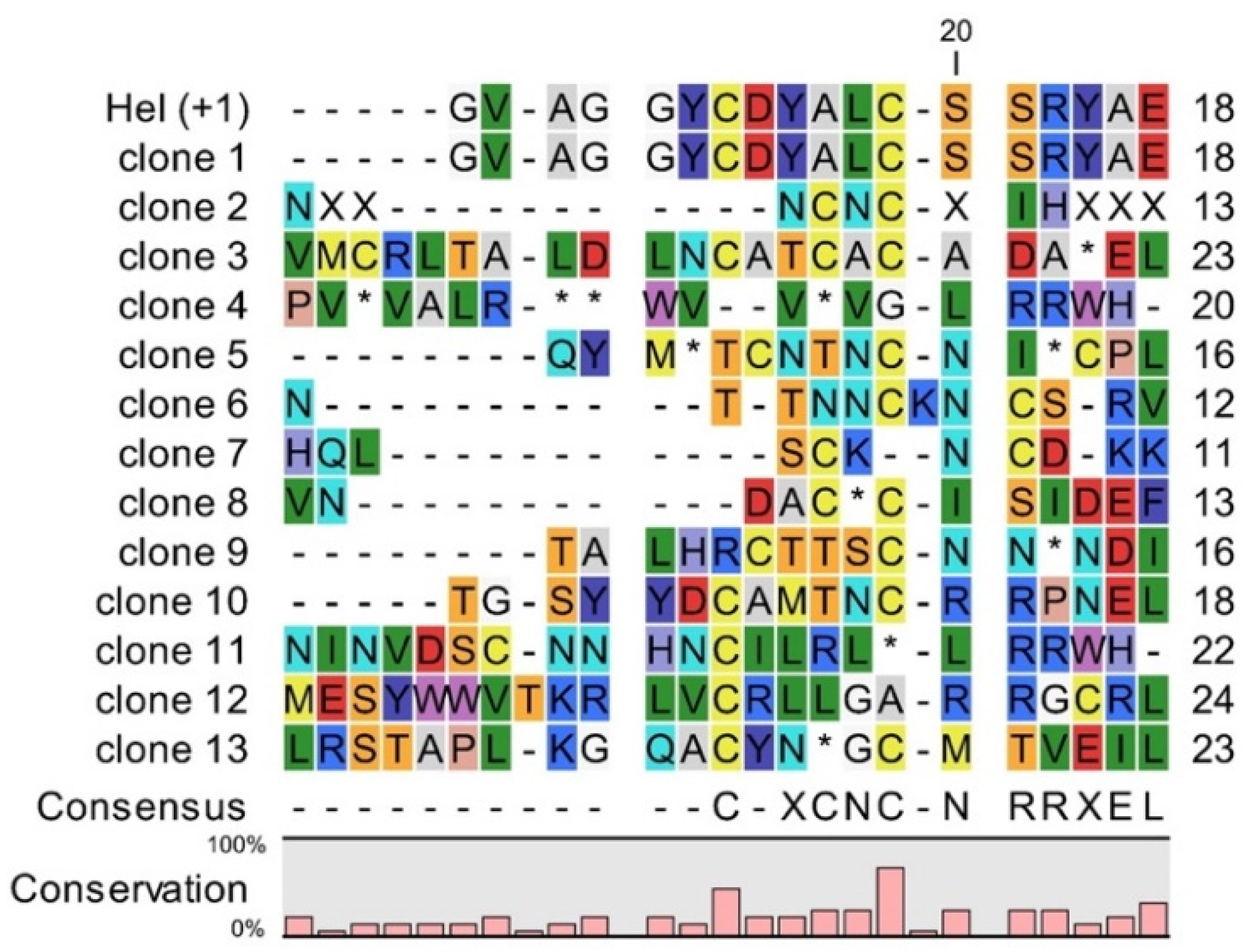
4.3. Sequencing of Selected Clones from vNAR Library with Randomized CDR3
4.4. Enrichment of Panning Round
4.5. Monoclonal Selection of Antibody Fragments
5. Reagents Setup
- Kanamycin solution: Dissolve kanamycin powder at 50 mg/mL in deionized water. Filterthrough 0.2 µm filter. Aliquot in 1 mL portions. Can be stored at −20 °C.
- Ampicillin solution: Dissolve ampicillin powder at 100 mg/mL in deionized water. Filterthrough 0.2 µm filter. Aliquot in 1 mL portions. Can be stored at −20 °C indefinitely. ThawedAliquots should be freshly diluted 1000-fold into medium or agar.
- Glucose solution (40%): Dissolve 400 g of glucose in 1 L of deionized water. Filter through0.2 µm filter. Can be stored at 4 °C for several months.
- 2X TY medium pH 7.0: 1.6% (w/v) tryptone, 1% (w/v) yeast extract, 0.5% (w/v) NaCl)
- 2X TY-GA: 2X TY, 100 mg/mL ampicillin, 100 mM glucose
- 2X TY-GA agar plates: Dissolve 10 g tryptone, 5 g yeast extract, and 10 g NaCl in 1L water. Add 15 g agar-agar and autoclave. Cool down below 50 °C, add 1 mL of filter-sterilized ampicillin (100 mg/mL) and 50 mL of 40% (w/v) filter sterilized glucose. Mix with gentle stirring and fill one-third of 100 mm or 150 mm diameter polystyrene Petri dishes. Keep the plates at 4 °C.
- SB medium pH 7.0: Dissolve 30 g tryptone, 20 g yeast extract and 10 g MOPS (3-[N-morpholino]–propanesulfic acid) in 1L water. Dissolve and autoclave.
- Minimal medium agar plate: Add 5.6 g 5X M9 salt and 15 g agar to 500 mL of deionized water and autoclave. When cooled to 50-45° C, add 1 mL of 1 M MgSO4 (autoclaved), 0.1 mL of 1 M CaSO4 (autoclaved), 5 mL of 40% glucose, and 0.25 mL of 1% thiamine HCl (filter-sterilized). Mix with gentle stirring and pour on polystyrene Petri dishes. Keep the plates at 4 °C.
- SOC medium: Add 20 g of tryptone, 5 g of yeast extract and 0.5 g of NaCl to 950 mL of deionized water. Shake well and add 10mL of a 250 mM KCl. Adjust the pH of the medium to 7.0. autoclave for 20 min. Add 5 mL of a sterile solution of 2 M MgCl2.
- Agarose electrophoresis gel: For 1% agarose gel, use 1 g of agarose and 100 mL TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA, pH 8.0). Change the amount of agarose as needed to make 1.5 or 2% gels.
- H2O/HEPES: Add 1 mL of sterile HEPES 1M to 1 L of autoclaved ultra-pure water.
- Glycerol/HEPES: Add 1 mL of sterile HEPES 1M to 1L 10% glycerol.
- TBSC buffer: 10 mM Tris pH 7.4, 137 mM NaCl, 1 mM CaCl2. Dissolve 1.5 g of TrisBase, 8 g NaCl and 0.15 g CaCl2 in 1 L of deionized water. Adjust to pH 7.4 and autoclave.
- PEG solution: 20% PEG, 2.5 M NaCl. Dissolve 100 g of PEG 8000 and 73 g of NaCl in 500 mL of deionized water. Filter through 0.2 µm filter.
- Trypsin solution: Dissolve trypsin powder at 10 mg/mL in TBSC (trypsin stock). Freeze in 20 µL aliquots in liquid nitrogen. This can be stored at −20 °C for several months. For the experiment, dissolve 100 µlL of trypsin stock in 10 mL of TBSC (trypsin solution).
- PBST: 1X PBS + 0.1% (v/v) Tween® 20 up to 1X PBS + 1% (v/v) Tween® 20.
- MPBST: 2% skimmed milk in PBST, prepare freshly.
- Panning block solution: MPBST or BSA-PBST: 1% (w/v) BSA in PBST, prepare freshly.
- Periplasmic buffer: 100 mM Tris; 0.5 M Sucrose; 1 mM EDTA; pH 8.0
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage Display: Concept, Innovations, Applications and Future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Caucheteur, D.; Robin, G.; Parez, V.; Martineau, P. Construction of Synthetic Antibody Libraries. Methods Mol. Biol. 2018, 1827, 93–108. [Google Scholar] [CrossRef]
- Ponsel, D.; Neugebauer, J.; Ladetzki-Baehs, K.; Tissot, K. High Affinity, Developability and Functional Size: The Holy Grail of Combinatorial Antibody Library Generation. Molecules 2011, 16, 3675–3700. [Google Scholar] [CrossRef]
- Paduch, M.; Koide, A.; Uysal, S.; Rizk, S.S.; Koide, S.; Kossiakoff, A.A. Generating Conformation-Specific Synthetic Antibodies to Trap Proteins in Selected Functional States. Methods 2013, 60, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Bian, H.; Wu, X.; Fu, T.; Fu, Y.; Hong, J.; Fleming, B.D.; Flajnik, M.F.; Ho, M. Construction and Next-Generation Sequencing Analysis of a Large Phage-Displayed VNAR Single-Domain Antibody Library from Six Naïve Nurse Sharks. Antib. Ther. 2019, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haußner, C.; Lach, J.; Eichler, J. Synthetic Antibody Mimics for the Inhibition of Protein–ligand Interactions. Curr. Opin. Chem. Biol. 2017, 40, 72–77. [Google Scholar] [CrossRef]
- Barelle, C.; Gill, D.S.; Charlton, K. Shark Novel Antigen Receptors—The Next Generation of Biologic Therapeutics? Adv. Exp. Med. Biol. 2009, 655, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Barelle, C.; Porter, A. VNARs: An Ancient and Unique Repertoire of Molecules That Deliver Small, Soluble, Stable and High Affinity Binders of Proteins. Antibodies 2015, 4, 240–258. [Google Scholar] [CrossRef]
- Zhao, A.; Tohidkia, M.R.; Siegel, D.L.; Coukos, G.; Omidi, Y. Phage Antibody Display Libraries: A Powerful Antibody Discovery Platform for Immunotherapy. Crit. Rev. Biotechnol. 2016, 36, 276–289. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Smith, G.P.; Scott, J.K. Libraries of Peptides and Proteins Displayed on Filamentous Phage. Methods Enzymol. 1993, 217, 228–257. [Google Scholar] [PubMed]
- Nelson, R.S.; Valadon, P. A Universal Phage Display System for the Seamless Construction of Fab Libraries. J. Immunol. Methods 2017, 450, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Kolmar, H.; Haberkorn, U.; Mier, W. DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods. Molecules 2011, 16, 1625–1641. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, T.M.; Yumerefendi, H.; Kuhlman, B.; Leaver-Fay, A. SwiftLib: Rapid Degenerate-Codon-Library Optimization through Dynamic Programming. Nucleic Acids Res. 2015, 43, e34. [Google Scholar] [CrossRef] [PubMed]
- Yagodkin, A.; Azhayev, A.; Roivainen, J.; Antopolsky, M.; Kayushin, A.; Korosteleva, M.; Miroshnikov, A.; Randolph, J.; Mackie, H. Improved Synthesis of Trinucleotide Phosphoramidites and Generation of Randomized Oligonucleotide Libraries. Nucleosides Nucleotides Nucleic Acids 2007, 26, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Van den Brulle, J.; Fischer, M.; Langmann, T.; Horn, G.; Waldmann, T.; Arnold, S.; Fuhrmann, M.; Schatz, O.; O’Connell, T.; O’Connell, D.; et al. A Novel Solid Phase Technology for High-Throughput Gene Synthesis. Biotechniques 2008, 45, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Stark, Y.; Venet, S.; Schmid, A. Whole Cell Panning with Phage Display. Methods Mol. Biol. 2017, 1575, 67–91. [Google Scholar] [CrossRef]
- de Bruin, R.; Spelt, K.; Mol, J.; Koes, R.; Quattrocchio, F. Selection of High-Affinity Phage Antibodies from Phage Display Libraries. Nat. Biotechnol. 1999, 17, 397–399. [Google Scholar] [CrossRef]
- Goemans, C.; Denoncin, K.; Collet, J.-F. Folding Mechanisms of Periplasmic Proteins. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1517–1528. [Google Scholar] [CrossRef]
- Hust, M.; Steinwand, M.; Al-Halabi, L.; Helmsing, S.; Schirrmann, T.; Dübel, S. Improved Microtitre Plate Production of Single Chain Fv Fragments in Escherichia Coli. Nat. Biotechnol. 2009, 25, 424–428. [Google Scholar] [CrossRef]
- Dooley, H.; Flajnik, M.F.; Porter, A.J. Selection and Characterization of Naturally Occurring Single-Domain (IgNAR) Antibody Fragments from Immunized Sharks by Phage Display. Mol. Immunol. 2003, 40, 25–33. [Google Scholar] [CrossRef]
- Shao, C.Y.; Secombes, C.J.; Porter, A.J. Rapid Isolation of IgNAR Variable Single-Domain Antibody Fragments from a Shark Synthetic Library. Mol. Immunol. 2007, 44, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Hust, M.; Mersmann, M. Phage Display and Selection in Microtitre Plates. In Antibody Engineering; Springer: Berlin/Heidelberg, Germany, 2010; pp. 139–149. [Google Scholar]
- Sidhu, S.S.; Lowman, H.B.; Cunningham, B.C.; Wells, J.A. Phage Display for Selection of Novel Binding Peptides. Methods Enzymol. 2000, 328, 333–363. [Google Scholar] [CrossRef] [PubMed]
- Mersmann, M.; Schmidt, A.; Tesar, M.; Schoneberg, A.; Welschof, M.; Kipriyanov, S.; Terness, P.; Little, M.; Pfizenmaier, K.; Moosmayer, D. Monitoring of ScFv Selected by Phage Display Using Detection of ScFv—PIII Fusion Proteins in a Microtiter Scale Assay. J. Immunol. Methods 1998, 220, 51–58. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) |
|---|---|
| ForFR1_NcoI | GCATGACCATGGCTCGAGTGGACCAAACACCG |
| RevFR4_NotI | GCATGAGCGGCCGCTTTATTCACAGTCACGGCAGTGCCAT |
| For_pSex | GTATGTTGTGTGGAATTGTG |
| Rev_pSex | GTTTTGTCGTCTTTCCAG |
| RevRan_12 | CGGCAGTGCCATCTCCGCA(MNN)3GCA(MNN)2-ACA(MNN)5GAGACCGCAACGATACGTGCCAC |
| RevRan_13 | CGGCAGTGCCATCTCCGCA(MNN)6GCA(MNN)1- ACA(MNN)4GAGACCGCAACGATACGTGCCAC |
| RevRan_16 | CGGCAGTGCCATCTCCGCA(MNN)6GCA(MNN)3- ACA(MNN)5GAGACCGCAACGATACGTGCCAC |
| RevRan_18 | CGGCAGTGCCATCTCCGCA(MNN)6GCA(MNN)4- ACA(MNN)6GAGACCGCAACGATACGTGCCAC |
| RevRan_20 | CGGCAGTGCCATCTCCGCA(MNN)5GCA(MNN)5- ACA(MNN)8GAGACCGCAACGATACGTGCCAC |
| RevRan_23 | CGGCAGTGCCATCTCCGCA(MNN)6GCA(MNN)5- ACA(MNN)10GAGACCGCAACGATACGTGCCAC |
| Elution Method | Readout | |
|---|---|---|
| Solutions Used | Time (min) | Normalized Phage Titer |
| solution A | 30 | 32× |
| solution B | 10 | 3× |
| solution C | 10 | 1× |
| solution A + solution B | 30 + 10 | 30× |
| solution B (two times) | 10 + 10 | 18× |
| solution A (two times) | 30 + 20 | 50× |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solemani Zadeh, A.; Grässer, A.; Dinter, H.; Hermes, M.; Schindowski, K. Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries. Methods Protoc. 2019, 2, 17. https://doi.org/10.3390/mps2010017
Solemani Zadeh A, Grässer A, Dinter H, Hermes M, Schindowski K. Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries. Methods and Protocols. 2019; 2(1):17. https://doi.org/10.3390/mps2010017
Chicago/Turabian StyleSolemani Zadeh, Arghavan, Alissa Grässer, Heiko Dinter, Maximilian Hermes, and Katharina Schindowski. 2019. "Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries" Methods and Protocols 2, no. 1: 17. https://doi.org/10.3390/mps2010017
APA StyleSolemani Zadeh, A., Grässer, A., Dinter, H., Hermes, M., & Schindowski, K. (2019). Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries. Methods and Protocols, 2(1), 17. https://doi.org/10.3390/mps2010017