Efficient Incorporation of Unnatural Amino Acids into Proteins with a Robust Cell-Free System


Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.1.1. Preparation of E. coli Extract
- E. coli Rosetta (DE3) strain (Biomed, Beijing, China; Cat. no.: BC204-01)
- Antifoam 204 (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: SLBW1473)
- Chloramphenicol (Genview, Beijing, China; Cat. no.: AC060)
- Tryptone (OXIOD, Basingstoke, UK; Cat. no.: LP0042)
- Yeast extract (OXIOD; Cat. no.: LP0021)
- BactoTM agar (Becton. Dickinson, Franklin Lakes, NJ, USA; Cat. no.: 7291815)
- NaCl (Sinopharm Chemical Reagent, Shanghai, China; Cat. no.: 10019318)
- Dipotassium hydrogen phosphate (KH2PO4) (Tong Guang Fine Chemicals, Beijing, China; Cat. no.: 7778-77-0)
- Potassium hydrogen phosphate trihydrate (K2HPO4·3H2O) (Tong Guang Fine Chemicals; Cat. no.: 16788-57-1)
- Ampicillin (Solarbio, Beijing, China; Cat. no.: A8180)
- Potassium L-glutamate (Yuanye, Shanghai, China; Cat. no.: S20427)
- L-Glutamic acid hemimagnesium salt tetrahydrate (Sigma-Aldrich; Cat. no.: 49605)
- Tris (Biotopped, Beijing, China; Cat. no.: T6061)
- 1,4-Dithio-DL-threitol (DTT) (Solarbio; Cat. no.: D8220)
2.1.2. Preparation of pPaFRS
- The nucleotide sequence of pPaFRS refers to pPRMjRS-1 [18]
- Plasmid pEVOL-pAzF [19] (pEVOL-pAzF was a gift from Peter Schultz (Addgene plasmid #31186; http://n2t.net/addgene:31186; RRID: Addgene_31186))
- Plasmid pET24a (Novagen, Shanghai, China; Cat. no.: 69749-3)
- Primers P1f, P1r, P2f, P2r, P3f and P3r (See Table S1)
- Q5® High-Fidelity DNA Polymerases (New England Biolabs, Beijing, China; Cat. no.: M0491)
- E. coli strain expressing the pPaFRS gene: BL21(DE3) competent cells (Biomed; Cat. no.: BC201)
- Kanamycin (Solarbio; Cat. no.: K8020)
- Tryptone (OXIOD; Cat. no.: LP0042)
- Yeast extract (OXIOD; Cat. no.: LP0021)
- BactoTM agar (Becton. Dickinson; Cat. no.: 7291815)
- NaCl (Sinopharm Chemical Reagent; Cat. no.: 10019318)
- Isopropyl-b-D-thiogalactoside (Solarbio; Cat. no.: I8070)
- EzFast Ni HP) columns (5 mL) (BestChrom, Shanghai, China; Cat. no.: EA005)
- Ethanol (TONG GUANG FINE CHEMICALS; Cat. no.: 32061)
- Imidazole (Sigma-Aldrich; Cat. no.: I2399)
- Quick Start Bradford Protein Assay Kit (Bio-Rad, Hercules, CA, USA; Cat. No.: 5000201)
- PBS buffer (Solarbio; Cat. no.: P1010)
2.1.3. Preparation of Crude T7 RNA Polymerase
- Plasmid pAR1219 (Sigma-Aldrich; Cat. no.: T2076).
- E. coli strain expressing the T7 RNA polymerase gene: BL21(DE3) competent cells (Biomed; Cat. no.: BC201)
- Tryptone (OXIOD; Cat. no.: LP0042)
- Yeast extract (OXIOD; Cat. no.: LP0021)
- BactoTM agar (Becton. Dickinson; Cat. no.: 7291815)
- NaCl (Sinopharm Chemical Reagent; Cat. no.: 10019318)
- Potassium acetate (Sigma-Aldrich; Cat. no.: V900213)
- Magnesium acetate tetrahydrate (Sigma-Aldrich; Cat. no.: V900172)
- Ethylenediaminetetraacetic acid (EDTA) (Solarbio; Cat. no.: E8040)
- 1,4-Dithio-DL-threitol (DTT) (Solarbio; Cat. no.: D8220)
- Potassium hydrogen phosphate trihydrate (K2HPO4·3H2O) (Tong Guang Fine Chemicals; Cat. no.: 16788-57-1)
- β-mercaptoethanol (Amresco, Shanghai, China; Cat. no.: 0482)
- 1 × Protease inhibitor (Sigma-Aldrich; Cat. no.: P8340)
2.1.4. Preparation of Expression Template and o-tDNAopt
- Plasmid pET23a (Novagen; Cat. no.: 69745-3)
- Primers P4f and P4r (See Table S1) to generate pET23a-sfGFP-StrepII gene.
- Primers P5f and P5r (See Table S1) to generate pET23a-sfGFP(2TAG)-StrepII gene which contained the TAG site in the 2nd codon and C-terminal StrepII tag.
- QIAGEN Plasmid Maxi Kit (10) (QIAGEN, Shanghai, China; Cat. no.: 12162)
- Plasmid Mini Kit (OMEGA Bio-Tek, Atlanta, GA, USA)
- The o-tDNAopt gene (GENEWIZ, Suzhou, China)
- Primers P6f and P6r to amplify the pET23a vector gene (See Table S1)
- Pfu polymerase (Beyotime Biotechnology, Shanghai, China; Cat. no.: D7217)
- Ethanol (Tong Guang Fine Chemicals; Cat. no.: 32061)
- 3 M Sodium acetate (Solarbio; Cat. no.: A1070)
2.1.5. Synthesis and Characterization of sfGFP and sfGFP2pPaF
- 10 × salt: 1.75 M Potassium glutamate, 27 mM Potassium oxalate monohydrate, 100 mM Glutamate, adjusted the pH to 7.2~7.4 with ammonia while dissolving
- Mg2+ solution: 1 M Magnesium glutamate.
- Amino acids mixture: Added the amino acids in the following order (given in three-letter code): Arg, Val, Trp, Phe, Ile, Leu, Cys, Met, Ala, Asn, Asp, Gly, Gln, His, Lys, Pro, Ser, Thr, Tyr. During preparation, it was necessary to ensure that any of amino acids has been dissolved before adding the next, and Tyr was added at last. Adjusted the pH to 7.4 with ammonia hydroxide.
![Mps 02 00016 i001]() CRITICAL STEP Phosphoenolpyruvic acid (PEP) solution: (Prepare rapidly on the ice and flash frozen) 883 mM Phosphoenolpyruvate, added 10 M KOH to adjust pH to 7.4.
CRITICAL STEP Phosphoenolpyruvic acid (PEP) solution: (Prepare rapidly on the ice and flash frozen) 883 mM Phosphoenolpyruvate, added 10 M KOH to adjust pH to 7.4.- GSSG (Oxidized glutathione) and GSH (Reduced glutathione): 100 mM GSSG, 100 mM GSH.
- 25 × NTPs mixture: Prepared and added all reagents one by one as the following order: 1 M putrescine, 1.5 M spermidine, 8.3 mM NAD, 30 mM ATP, 21.5 mM CTP, GTP and UTP, 6.8 mM CoA, 4.3 mg/mL E. coli tRNA, and 0.9 mg/mL folinic acid. Before adding the next reagent, ensure the last reagent was completely dissolved. The final pH should be between 7.4 and 7.6. Store at −80 °C.
- pPaF solution: 100 mM (Medchem Source LLP, Washington, America; Cat. no.: JA-1003).
- E. coli extract (Prepared in 3.1).
- The pPaFRS (Prepared in 3.2).
- 25 mM of DTT.
- 55 mM iodoacetamide.
- 0.1% trifluoroacetic acid in 50% acetonitrile aqueous solution.
- Sequencing grade modified trypsin (Promega, America; Cat. No. #V5117).
- Mobile phase A: 0.1% formic acid.
- Mobile phase B: 100% acetonitrile and 0.1% formic acid.

2.2. Equipment
2.2.1. Preparation of E. coli Extract
- 1 L flasks.
- Constant temperature shaker.
- BIOSTAT® A plus Bioreactor (Sartorius, Gottingen, Germany)
- Ultrospec 3100 pro UV/Visible spectrophotometer (Amersham, Piscataway, NJ, USA)
- Micro Refrigerated Centrifuge Model 3700 (KUBOTA, Osaka, Japan)
- Vortex-Genie 2 (Scientific Industries, Bohemia, NY, USA)
- JN-3000PLUS high press crusher (JNBIO, China)
- Spectra/Por #1 dialysis tubing, MWCO 6-8 kD (Spectrum Laboratories, Rancho Dominguez, CA, USA)
- MS-H-Pro+ magnetic stirring apparatus (DRAGONLAB, Beijing, China)
2.2.2. Preparation of pPaFRS
- 1 L flasks.
- Constant temperature shaker.
- JN-3000PLUS high press crusher (JNBIO)
- Ultrospec 3100 pro UV/Visible spectrophotometer (Amersham)
- Micro Refrigerated Centrifuge Model 3700 (KUBOTA)
- Vortex-Genie 2 (Scientific Industries)
- ÄKTAprime plus (GE Healthcare, Chicago, IL, USA)
- MS-H-Pro+ magnetic stirring apparatus (DRAGONLAB)
- Amicon Ultra 15 mL 10 K (Merck Millipore, Darmstadt, Germany; Cat. No. UFC901096)
- Orbital Shaker TS-2 (Kylin-Bell Lab Instruments, Haimen, China)
2.2.3. Preparation of Crude T7 RNA Polymerase
- 1 L flasks.
- Constant temperature shaker.
- Ultrospec 3100 pro UV/Visible spectrophotometer (Amersham)
- Micro Refrigerated Centrifuge Model 3700 (KUBOTA)
- Vortex-Genie 2 (Scientific Industries)
- Qsonica Q700 Ultrasonic crusher (Misonix, Farmingdale, NY, USA)
- MS-H-Pro+ magnetic stirring apparatus (DRAGONLAB)
2.2.4. Preparation of Expression Template and o-tDNAopt
- Constant temperature shaker.
- Micro Refrigerated Centrifuge Model 3700 (KUBOTA)
- Vortex-Genie 2 (Scientific Industries)
- Plasmid mini kit (Omega Bio-Tek)
- C100TM Thermal Cycler (Bio-Rad)
- NanoVue Plus (GE Healthcare)
2.2.5. Synthesis and Characterization of sfGFP and sfGFP2pPaF
- Infinite M200 PRO Microplate reader (Tecan, Switzerland)
- Strep-Tactin affinity chromatography (IBA GmbH, Goettingen, Germany)
- Amicon Ultra 15 mL 10 K (Merck Millipore; Cat. No. UFC901096)
- Mini-PROTEAN® Tetra Cell (Bio-rad)
- SpeedVac (Thermo Fisher Scientific, Waltham, MA, USA)
- EASY-nLC 1000 system (Thermo Fisher Scientific)
- Analytical column: A home-made fused silica capillary column (75 µm ID, 150 mm length; Upchurch, Oak Harbor, WA, USA) packed with C-18 resin (300 Å, 5 µm, Varian, Lexington, MA)
- Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany)
- Xcalibur3.0 software
- Proteome Discoverer (Version PD1.4, Thermo Fisher Scientific)
3. Procedure
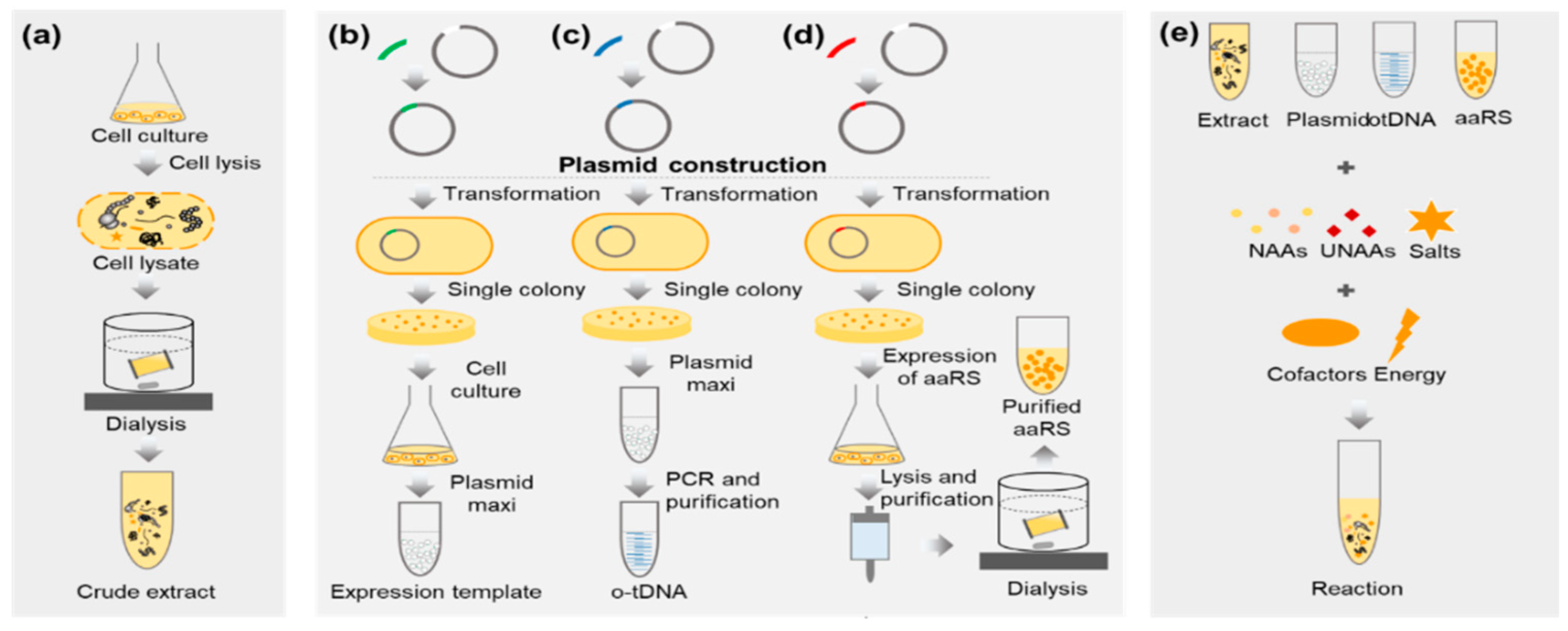
3.1. Preparation of E. coli Extract (Time for Completion: 2 Days)
- Prepared solutions used in this part.
- (1)
- 2 × Yeast extract-Tryptone (YT)-Phosphate (P)-Chloramphenicol (Cm) medium (10 g/L Yeast extract, 16 g/L Tryptone, 5 g/L NaCl, 40 mM K2HPO4, 22 mM KH2PO4, 34 mg/L chloramphenicol, and 1.5% agar for plate use if needed).
- (2)
- S30 buffer A: 14 mM L-Glutamic acid hemimagnesium salt tetrahydrate, 60 mM Potassium L-glutamate, 50 mM Tris, pH 7.7, titrated with acetic acid. Add DTT to 2 mM just before use. Stored at 4 °C.
- (3)
- S30 buffer B: 14 mM L-Glutamic acid hemimagnesium salt tetrahydrate, 60 mM Potassium L-glutamate, pH 8.2, titrated with Tris. Add DTT to 1 mM just before use. Stored at 4 °C.
- Prepared and autoclaved 2 × YT-P medium (10 mL, 200 mL, and 4 L in the bioreactor) and S30 A buffer (500 mL) and S30 B buffer (2 L).
- Cultivated E. coli Rosetta(DE3) strain on 2 × YT-P-Cm solid medium, and incubated overnight at 37 °C.
- Selected single colony and transferred it to 10 mL liquid 2xYT-P medium in 50 mL flask with Cm and incubated overnight at 37 °C with 220 rpm.
- Transferred 10 mL overnight culture into 200 mL fresh 2xYT-P medium in 1 L flask and continued culturing for about 2 h in the shaker.
- When the OD600 of culture reached to 2 to 3, it was transferred into a 4-L bioreactor (Sartorius) with Cm and 400 μL antifoam. Controlled the fermentation conditions at 37 °C and 500 rpm stirring.
![Mps 02 00016 i001]() CRITICAL STEP When the OD600 reached to 3.5 to 4.0, harvested the cells quickly at 4 °C to obtain high activity of cell extracts.
CRITICAL STEP When the OD600 reached to 3.5 to 4.0, harvested the cells quickly at 4 °C to obtain high activity of cell extracts.![Mps 02 00016 i002]() PAUSE STEP Washed the cell pellets with 100 mL S30 buffer A at least twice (after being washed, the pellet could be stored at 4 °C overnight or at −80 °C for a long time).
PAUSE STEP Washed the cell pellets with 100 mL S30 buffer A at least twice (after being washed, the pellet could be stored at 4 °C overnight or at −80 °C for a long time).- Re-suspended the pellets in 1 mL of S30 buffer A per gram of biomass on the ice.
- Subjected the suspension to a high press crusher (JNBIO) twice at 15000~20000 psi.
- Centrifuged the lysed cells at 4 °C and 13,000× g for at least 30 min.
- Incubated the supernatant at 37 °C with 120 rpm for 80 min.
- Centrifuged the extract at 4 °C and 13,000× g for at least 30 min.
- Transferred the supernatant to 6–8 kDa MWCO dialysis tubing and did dialysis in 100 times volume of supernatant S30 buffer B overnight at 4 °C (or 4 h twice).
- Re-centrifuged the extract at 4 °C with 13,000× g for 30 min.
- Collected and transferred the supernatant to 1.5 mL Eppendorf tubes on ice.
- Flash-frozen the extracts in liquid nitrogen and stored them at −80 °C.

3.2. Preparation of pPaFRS (Time for Completion: 5~6 Days from Plasmid Construction to pPaFRS Purification)
- Prepared buffers used in this part.
- (1)
- His-tag binding buffer: 30 mM Imidazole, 20 mM Na3PO4, 500 mM NaCl, titrated with phosphoric acid to pH 7.4. Stored at 4 °C.
- (2)
- His-tag elution buffer: 500 mM Imidazole, 20 mM Na3PO4, 500 mM NaCl, titrated with phosphoric acid to pH 7.4. Stored at 4 °C.
- Amplified the pET24a vector and pPaFRS gene by Q5® High-Fidelity DNA Polymerases system.
- Ligated the pET24a vector and pPaFRS gene with the homologous arm at 50 °C and 15 min.
- Screened with LB-K plate (LB plate with 50 μg/mL Kanamycin) by DH5α.
- Selected a single colony and sequenced it.
- Transformed pET24a-6H-pPaFRS into E. coli BL21(DE3), spread the cell on a LB-K plate, and incubated overnight at 37 °C.
- Picked up a single colony with a toothpick and inoculated it directly into 10 mL of LB-K medium followed by incubation for 12 h at 37 °C and 220 rpm.
- Transferred 10 mL culture into 200 mL LB-K medium and continued culturing it overnight in a shaker.
- Transferred the cells into 1 L fresh LB-K medium at a 5% inoculation amount.
- When the OD600 reached 0.6–0.8, added 1 mM of Isopropyl β-D-Thiogalactoside (IPTG).
- The cells were further cultured for 3–4 h at 37 °C. Cultivated the cells at 4 °C and 10,000 × g.
![Mps 02 00016 i002]() PAUSE STEP Washed the cells with 100 mL His-tag binding buffer at least twice (after being washed, the pellets could be stored at 4 °C overnight or at −80 °C for a long time).
PAUSE STEP Washed the cells with 100 mL His-tag binding buffer at least twice (after being washed, the pellets could be stored at 4 °C overnight or at −80 °C for a long time).- Re-suspended the pellets with suitable His-tag binding buffer, making the final optical density at a wavelength of 600 nm (OD600) between 40 and 60 on ice.
- Subjected the suspension to high press crusher single time at 15,000~20,000 psi.
- Centrifuged the lysate at 4 °C and 13,000 × g for 30 min.
- Filtrated the lysate with 0.45 μm water filters.
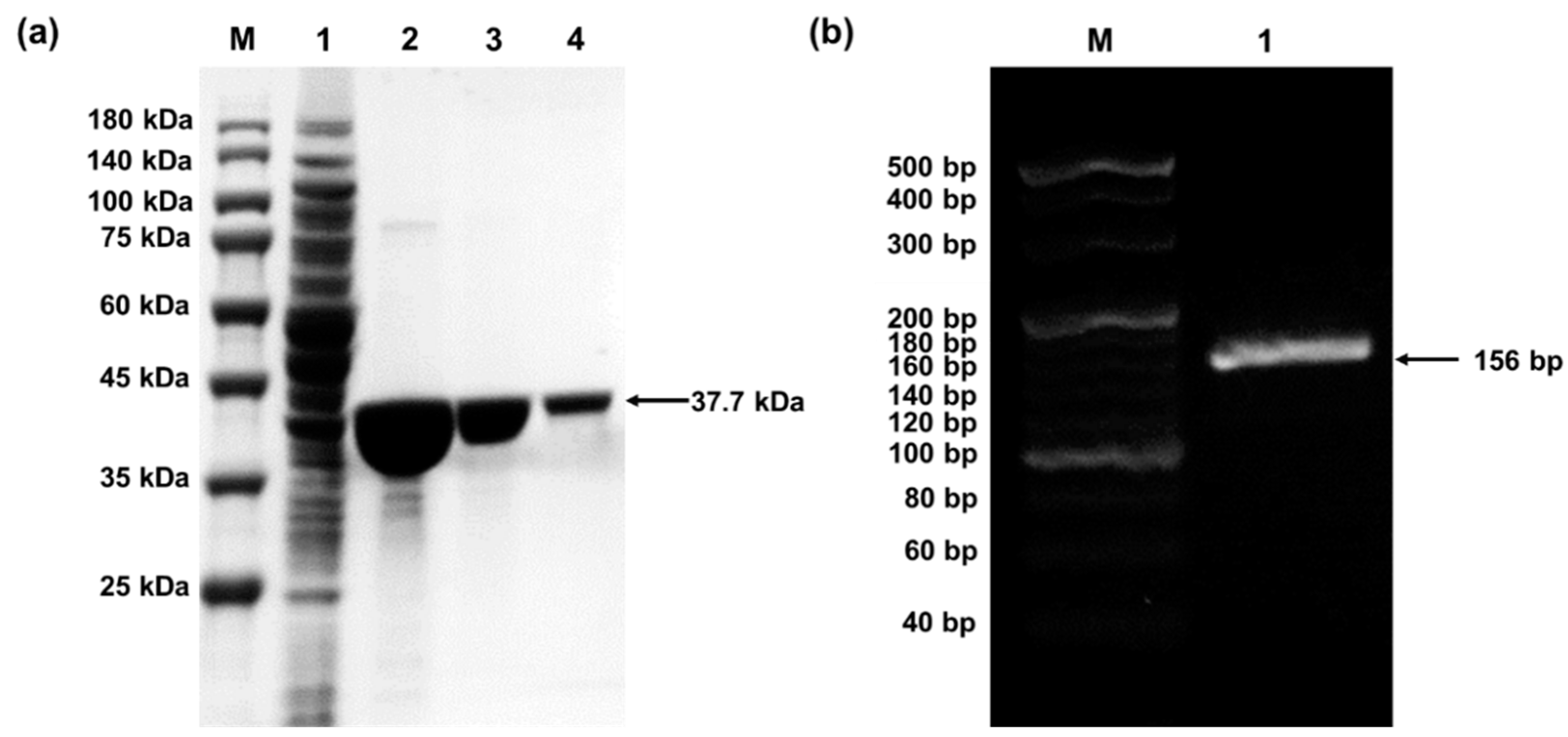
![Mps 02 00016 i001]() CRITICAL STEP All the lysate was loaded onto a 5 mL EzFast Ni HP column, which was connected with the ÄKTA Prime system and equilibrated with His-tag binding buffer. Then, the target pPaFRS was eluted with His-tag elution buffer and collected eluate in 1 mL fractions (all buffers should eliminate bubbles and be stored at 4 °C).
CRITICAL STEP All the lysate was loaded onto a 5 mL EzFast Ni HP column, which was connected with the ÄKTA Prime system and equilibrated with His-tag binding buffer. Then, the target pPaFRS was eluted with His-tag elution buffer and collected eluate in 1 mL fractions (all buffers should eliminate bubbles and be stored at 4 °C).- Placed the eluate in the 6–8 kDa MWCO dialysis tubing and dialyzed it against 50–100 volumes of sterile PBS (pH 7.4) buffer overnight.
- Determined protein concentrations using Quick Start Bradford Protein Assay Kit. When necessary, the fractions were concentrated using Amicon Ultra centrifugal device (10 kDa).
- Added 20% (v/v%) sucrose to fractions, and stored at −80 °C.
3.3. Preparation of Crude T7 RNA Polymerase (Time for completion: 3 days)
- Prepared buffers used in this part (stored at 4 °C).
- (1)
- Lysis buffer: 50 mM NaCl, 10 mM EDTA, 10 mM K2HPO4, 1 mM DTT, 10 mM β- mercaptoethanol, 1 × Protease inhibitor, 5% glycerin, pH 8.0.
- (2)
- Dialysis buffer: 50 mM NaCl, 1 mM EDTA, 40 mM K2HPO4, 1 mM DTT, 20% Sucrose, pH 7.7.
- Transformed E. coli BL21(DE3) with pAR1219, and incubated it on LB plate with 100 μg/mL Ampicillin (LB-A) overnight at 37 °C.
- Selected a single colony to inoculate in 10 mL of LB-A medium and cultured cells for 12 h at 37 °C and 220 rpm.
- Transferred 10 mL culture into 100 mL LB-A medium and continued culturing cells overnight in a shaker.
- Transferred cells into fresh LB-A medium at a 5% inoculation amount.
- When OD600 reached 0.6–0.8, IPTG was added to a final concentration of 0.1 mM.
- When OD600 reached 2.0, cells were harvested for 10,000 × g at 4 °C.
![Mps 02 00016 i002]() PAUSE STEP The cells were washed with 5 mL ice-cold wash buffer per gram pellet for twice (after washing, the pellet can be stored at 4 °C overnight or at −80 °C for a long time).
PAUSE STEP The cells were washed with 5 mL ice-cold wash buffer per gram pellet for twice (after washing, the pellet can be stored at 4 °C overnight or at −80 °C for a long time).- Re-suspended in 4 mL ice-cold lysis buffer per gram cells.
![Mps 02 00016 i001]() CRITICAL STEP Cells were ultrasonicated for 40 min on the ice working for two seconds and intermittent for two seconds (Kept the sample on the ice to maintain the activity).
CRITICAL STEP Cells were ultrasonicated for 40 min on the ice working for two seconds and intermittent for two seconds (Kept the sample on the ice to maintain the activity).- Cells were centrifuged at 13,000 × g and 4 °C for 20 min and discarded cell pellet.
- Dialyzed (6–8 kDa) twice in the dialysis buffer (100 times volume of samples) at 4 °C overnight. The suspension was centrifuged at 10,000× g for 30 min at 4 °C and the pellet was discarded.
- The crude T7 RNA polymerase was flash-frozen in liquid nitrogen, and stored at −80 °C until use.
3.4. Preparation of Expression Template and o-tDNAopt (Time for Completion: 2 Days)
- Made pET23a-sfGFP-StrepII and pET23a-sfGFP(2TAG)-StrepII plasmids self-ligated by homology arms at 50 °C at least 15 min.
- Added the ligation system into 100 μL DH5α competent cells and put them on ice for 30 min.
- Added LB medium about 400 μL and cultured them at 37 °C and 220 rpm about 30 min.
- Coated about 100 μL suspension on the LB-A plate.
- Picked up two single colonies to sequence.
- Selected the correct strain and extracted the plasmid with QIAGEN Plasmid Maxi Kit.
- Ligated the o-tDNAopt and the pET23a vector genes at 50 °C at least 15 min.
- Screening method was the same as 3.4 2–5.
- Extracted the pET23a o-tDNAopt with Plasmid Mini Kit as an o-tDNAopt amplification template.
- Amplified o-tDNA gene with Pfu polymerase.
- Purified o-tDNA gene by ethanol precipitation.
- The o-tDNAopt was diluted with MiliQ water to 2 mg/mL and stored at –20 °C.
3.5. Synthesis and Characterization of sfGFP and sfGFP2pPaF (Time for Completion: 3~4 Days)
- Thawed the CFPS, OTS components, and expression templates on the ice.
- The standard 20 μL cell-free reaction mixture consisted of the followings in Table 1.
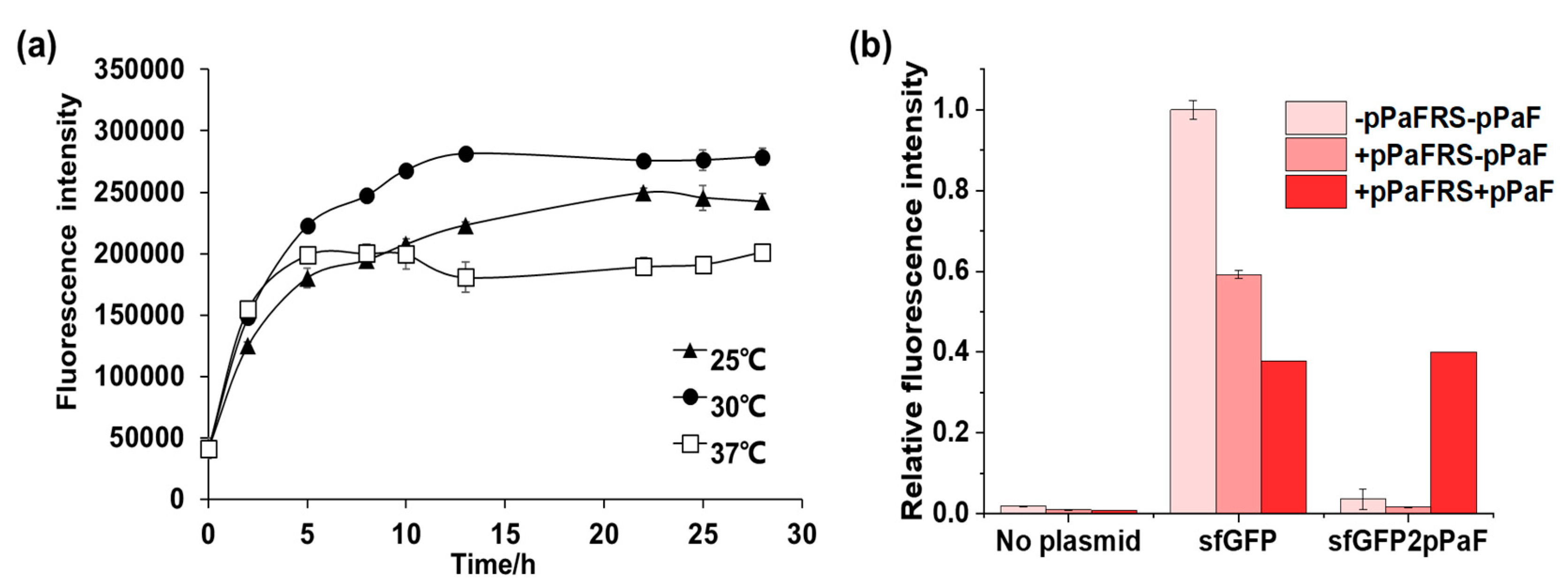
- Mixed the mixture and reacted at 30 °C for 16 h.
- Five-microliter samples from each reaction mixture were diluted with 195 μL ddH2O, and the fluorescence intensity of these diluted samples was measured with F485 excitation and F535 emission filters using Microplate reader.
- Preparation of samples for mass spectrometry:
- (1)
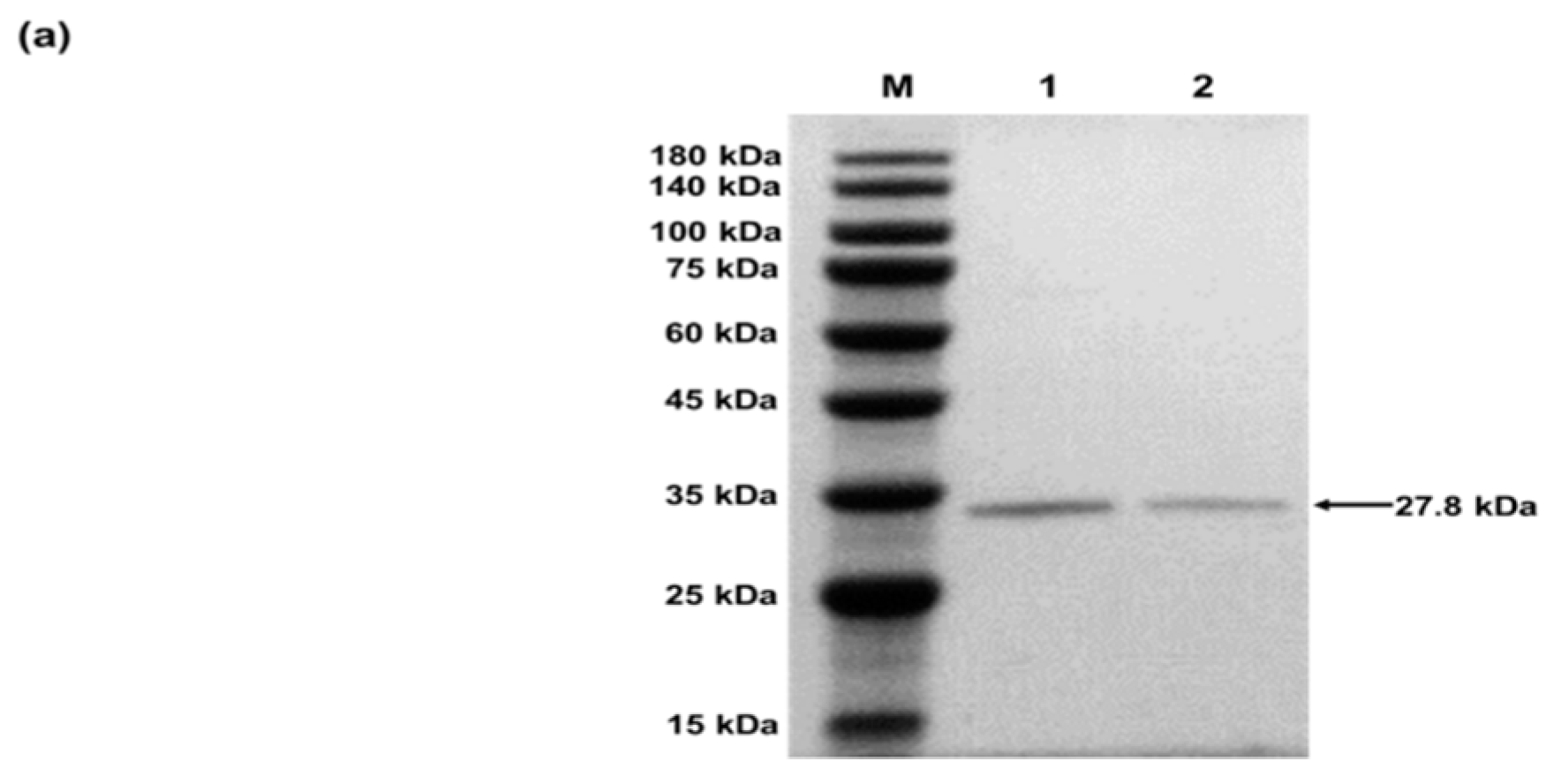
- Purified sfGFP and sfGFP2pPaF produced in CFPS with a C-terminal Strep-tag via Strep-Tactin affinity chromatography according to the manufacturer instructions.
- (2)
- Dialyzed and concentrated the fractions obtained from Strep-Tactin affinity chromatography with PBS (pH 7.4) buffer at 4 °C.
- (3)
- Determined protein concentrations using Quick Start Bradford Protein Assay Kit.
- (4)
- Analyzed the samples by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
- (5)
- Extracted interested bands from the gel.
- (6)
- Reduced with 25 mM of DTT and alkylated with 55 mM iodoacetamide and digested in gel with sequencing grade modified trypsin at 37 °C overnight.
- (7)
- Extracted peptides twice with 0.1% trifluoroacetic acid in 50% acetonitrile aqueous solution for 30 min and then dried in a SpeedVac.
- (8)
- Redissolved peptides in 25 μL 0.1% trifluoroacetic acid.
- (9)
- Analyzed 6 μL of extracted peptides by Thermo orbitrap fusion.
- (10)
- LC-MS/MS analysis of samples was performed at the Center of Biomedical Analysis of Tsinghua University.
4. Expected Results
4.1. Preparation of pPaFRS and o-tDNAopt
4.2. Synthesis and Characterization of sfGFP and sfGFP2pPaF
4.2.1. Synthesis and Characterization of sfGFP and sfGFP2pPaF
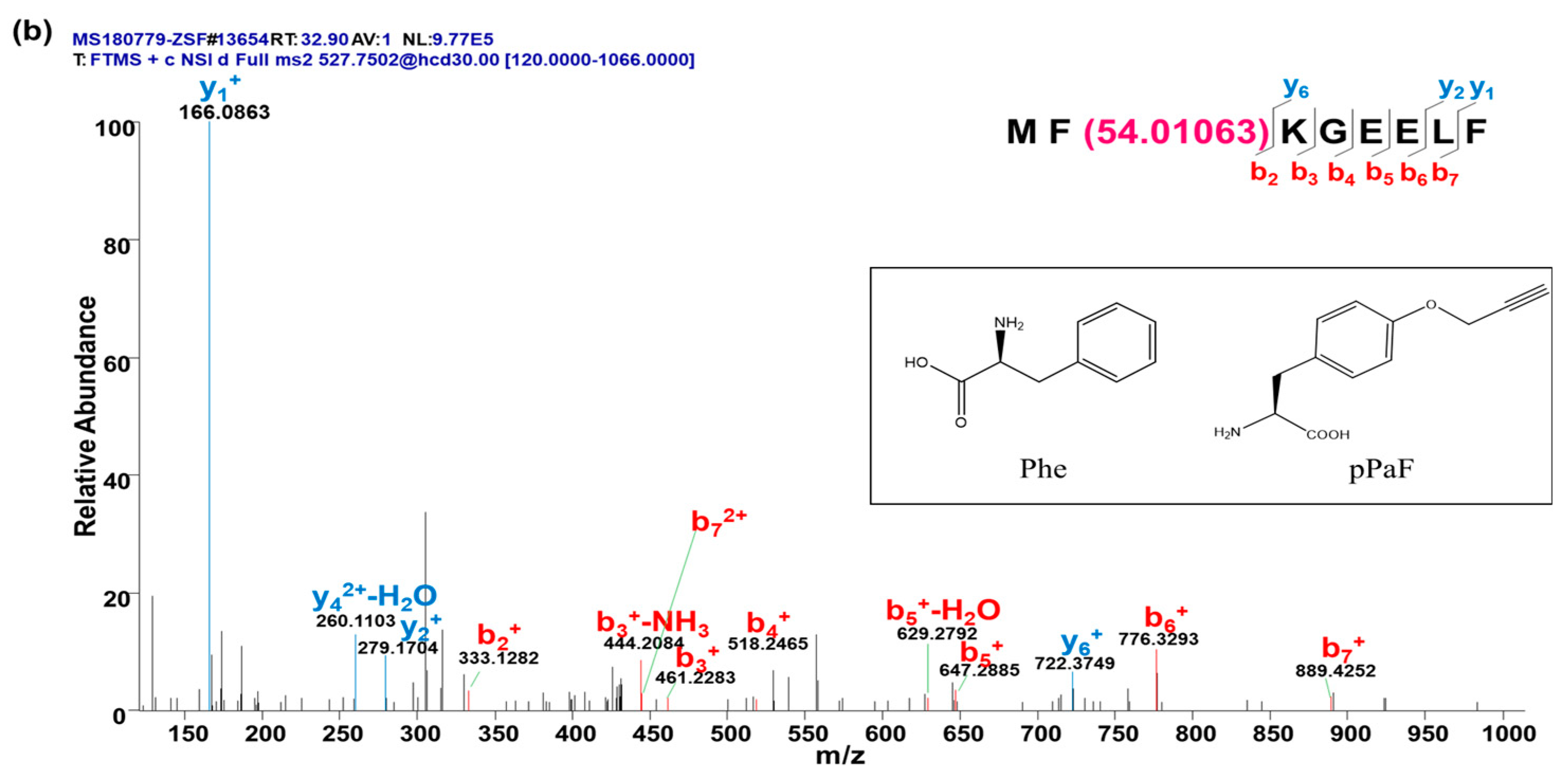
4.2.2. Characterization of sfGFP and sfGFP2pPaF
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soye, B.J.D.; Patel, J.R.; Isaacs, F.J.; Jewett, M.C. Repurposing the translation apparatus for synthetic biology. Curr. Opin. Chem. Biol. 2015, 28, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Lu, Y.Y.; Van Deventer, J.A.; Tirrell, D.A. Residue-specific incorporation of non-canonical amino acids into proteins: Recent developments and applications. Curr. Opin. Chem. Biol. 2010, 14, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Genetically encoding new bioreactivity. New Biotechnol. 2017, 38, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Leisle, L.; Valiyaveetil, F.; Mehl, R.A.; Ahern, C.A. Incorporation of non-canonical amino acids. Adv. Exp. Med. Biol. 2015, 869, 119–151. [Google Scholar] [PubMed]
- Quast, R.B.; Mrusek, D.; Hoffmeister, C.; Sonnabend, A.; Kubick, S. Cotranslational incorporation of non-standard amino acids using cell-free protein synthesis. FEBS Lett. 2015, 589, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Perez, J.G.; Carlson, E.D.; Ntai, I.; Isaacs, F.J.; Kelleher, N.L.; Jewett, M.C. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng. 2017, 114, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Kwon, Y.-C.; Jewett, M.C. Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, P.; Ling, J.; Wang, Y.-S.; Söll, D. Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 2013, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Bröcker, M.J.; Ho, J.M.L.; Church, G.M.; Söll, D.; O’Donoghue, P. Recoding the genetic code with selenocysteine. Angew. Chem. Int. Ed. 2014, 53, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Summerer, D.; Chen, S.; Wu, N.; Deiters, A.; Chin, J.W.; Schultz, P.G. A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. USA 2006, 103, 9785–9789. [Google Scholar] [CrossRef] [PubMed]
- Oza, J.P.; Aerni, H.R.; Pirman, N.L.; Barber, K.W.; ter Haar, C.M.; Rogulina, S.; Amrofell, M.B.; Isaacs, F.J.; Rinehart, J.; Jewett, M.C. Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun. 2015, 6, 8168. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, C.; Swartz, J.R. Direct polymerization of proteins. ACS Synth. Biol. 2014, 3, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Daniel, T.; Buechler, Y.J.; Litzinger, D.C.; Maio, Z.; Putnam, A.-M.H.; Kraynov, V.S.; Sim, B.-C.; Bussell, S.; Javahishvili, T.; et al. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2011, 108, 9060–9065. [Google Scholar] [CrossRef] [PubMed]
- Bundy, B.C.; Swartz, J.R. Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconj. Chem. 2010, 21, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y. Cell-free synthetic biology: Engineering in an open world. Synth. Syst. Biotechnol. 2017, 2, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.D.; Gan, R.; Hodgman, C.E.; Jewett, M.C. Cell-free protein synthesis: Applications come of age. Biotechnol. Adv. 2012, 30, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Caschera, F.; Noireaux, V. Synthesis of 2.3 mg/mL of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Schultz, P.G. In vivo incorporation of an alkyne into proteins in Escherichia coli. Bioorg. Med. Chem. Lett. 2005, 15, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Santoro, S.W.; Martin, A.B.; King, D.S.; Wang, L.; Schultz, P.G. Addition of p-azido-l-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, C.; Swartz, J.R. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013, 41, 5949–5963. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Volume (μL) |
|---|---|
| 10 × salt | 2 |
| PEP | 1.6 |
| Mg2+ | 0.4 |
| Extract | 5 |
| T7 polymerase | 0.2 |
| NTPs mixture | 0.8 |
| Amino acids mixture | 0.8 |
| Plasmid template | 1 (300 ng) |
| o-tDNA | 1 (100 ng/μL) |
| pPaFRS | 1 (5 mM) |
| ddH2O | 6.12 |
| Total | 20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, W.; Bu, N.; Lu, Y. Efficient Incorporation of Unnatural Amino Acids into Proteins with a Robust Cell-Free System. Methods Protoc. 2019, 2, 16. https://doi.org/10.3390/mps2010016
Gao W, Bu N, Lu Y. Efficient Incorporation of Unnatural Amino Acids into Proteins with a Robust Cell-Free System. Methods and Protocols. 2019; 2(1):16. https://doi.org/10.3390/mps2010016
Chicago/Turabian StyleGao, Wei, Ning Bu, and Yuan Lu. 2019. "Efficient Incorporation of Unnatural Amino Acids into Proteins with a Robust Cell-Free System" Methods and Protocols 2, no. 1: 16. https://doi.org/10.3390/mps2010016
APA StyleGao, W., Bu, N., & Lu, Y. (2019). Efficient Incorporation of Unnatural Amino Acids into Proteins with a Robust Cell-Free System. Methods and Protocols, 2(1), 16. https://doi.org/10.3390/mps2010016