High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates

 , , and
, , and
Abstract
1. Introduction
2. Experimental Design
2.1. Reagents and Materials
- Fetal bovine serum, FBS (Thermo Fisher, Paisley, UK; Cat. no.: 10270-106)
- Dimethyl sulphoxide, DMSO (VWR, Lutterworth, UK; Cat. no.: 23500.260)
- 96-well V-bottomed plates (Sigma-Aldrich, Dorset, UK; Cat. no.: CLS3894)
- Parafilm (VWR, Lutterworth, UK; Cat. no.: PM-996)
- 96-well PCR plate (Thermo Fisher; Cat. no.: AB1400L)
- Tris, 1 M, pH 8.0 (Thermo Fisher; Cat. no.: AM9855G)
- Ethylenediaminetetraacetic acid (EDTA), 0.5 mM, pH 8.0 (Thermo Fisher; Cat. no.: 15575-038)
- Tween 20, 50% (Thermo Fisher; Cat. no.: 003005)
- PCR grade water (Thermo Fisher; Cat. no.: AM9932)
- DNA low bind tubes (Eppendorf, Arlington, UK; Cat. no.: Z666548)
- Proteinase K (Thermo Fisher; Cat. no.: EO0491)
- Platinum PCR master mix (Thermo Fisher; Cat. no.: 12532016)
- Locus specific primers, 10 µM mix, see Section 2.3 (Sigma, St Louis, MO, USA or Integrated DNA Technologies (IDT), Skokie, Illinois, USA)
- Agarose (Roche, Burgess Hill, UK; Cat. no.: 11388983001)
- 10× Tris acetate-EDTA, TAE (Sigma-Aldrich; Cat. no.: 11666690001)
- 100 bp ladder (New England Biolabs, Hitchin, UK; Cat. no.: N0551G)
- Exonuclease I, Escherichia coli (New England Biolabs; Cat. no: M0293L)
- Shrimp alkaline phospatase, rSAP (New England Biolabs, Hitchin, UK; Cat. no.: M0371L)
- Custom iR5 and iC7 barcoded primers, Table 1 (Sigma or IDT)
- Agencourt Ampure XP SPRI Beads (Beckman Coulter, High Wycombe, UK; Cat. no.: A63881)
- 100% Ethanol (VWR, Lutterworth, UK; Cat. no.: 20821.330)
- Qubit BR DNA assay kit (Thermo Fisher; Cat. no.: Q32850)
- NEB Ultra II (New England Biolabs; Cat. no.: 7645S/L)
- PCR Tube (Appleton Woods, Birmingham, UK; Cat. no.: TA571)
- NEB Next Multiplex Oligos for Illumina primer set 1 (New England Biolabs; Cat. no.: E7500S/L)
- NEB Next Multiplex Oligos for Illumina primer set 2 (New England Biolabs; Cat. no.: E7335S/L)
- Herculase II Fusion polymerase kit (Agilent, Cheadle, UK; Cat. no.: 600677)
- Tris-EDTA, TE (Sigma-Aldrich; Cat. no.: 99302)
- D1000 reagents (Agilent; Cat. no.: 50675583)
- D1000 loading tips (Agilent; Cat. no.: 50675153)
- D1000 screen tape (Agilent; Cat. no.: 50675582)
- KAPA Library Quantification Complete Kit (Roche; Cat. no.: KK4824)
- MiSeq Reagent Nano Kit, v2 500-Cycles (Illumina, Cambridge, UK; Cat. no.: MS-103-1003)
- PhiX Control v3 (Illumina; Cat. no.: FC-110-3001)
2.2. Equipment
- 8- or 12-channel pipette (Labgene Scientific, Châtel-Saint-Denis, Switzerland: Cat. no.: 5121, 5125)
- Centrifuge with buckets for plates (Eppendorf; Cat. no.: 5810R)
- Thermocycler with 96-well plate capacity (Bio-Rad, Watford, UK; Cat. no.: T100)
- Electrophoresis gel tank and power pack
- Magnetic rack (DynaMag-2; Thermo Fisher; Cat. no.: 13221)
- Minifuge (Starlab, Milton Keynes, UK; Cat. no.: N2631-0007)
- Qubit fluorometer (Thermo Fisher; Cat. no.: Q33226)
- Agilent 2200 TapeStation (Agilent; Cat. no.: G2964AA)
- Real-time quantitative polymerase chain reaction (qPCR) thermocycler (Thermo Fisher StepOnePlus; Thermo Fisher; Cat. no.: 4376598)
- MiSeq (Illumina; Cat. no.: SY-410-1003)
- Microcentrifuge (Eppendorf; Cat. no.: 5424R)
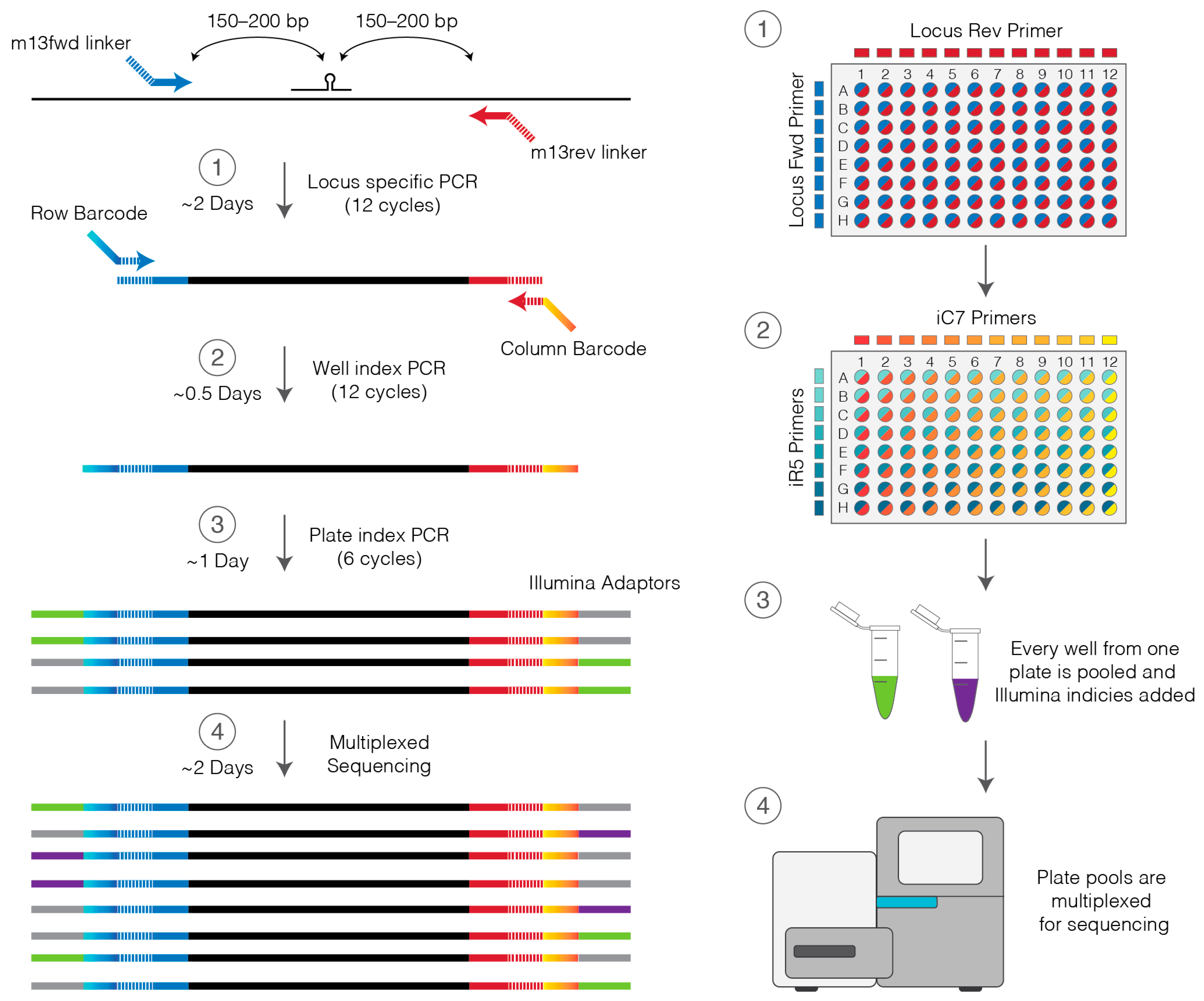
2.3. Custom Locus Primers
2.4. Primers to Barcode Individual Wells
2.5. Analysis Software
3. Procedure
3.1. Clonal Expansion. Time for Completion: 2–3 Weeks
- 1.
- Using the appropriate growth media, clonally expand single-cell sorted colonies, splitting them as necessary until they occupy two to four wells of a 96-well plate at a high level of confluence (80–90%).
3.2. Splitting and Freezing Cells. Time for Completion: 3 h
- 2.
- Reduce media volume to <100 µL per well and mix by pipetting.
- 3.
- Prepare two new 96-well V-bottomed plates (one for stock storage and one for genotyping) by combining two wells of highly confluent cells. Note: If cells have only grown to occupy two wells of a flat-bottomed 96-well plate, transfer a single well to each V-bottomed plate.
 CRITICAL STEP Clones should occupy identical wells in both the stock and genotyping plates.
CRITICAL STEP Clones should occupy identical wells in both the stock and genotyping plates.
- 4.
- Set aside the genotyping plate and pellet the cells in the stock plate by centrifugation (250× g, 5 min, room temperature).
- 5.
- During this spin step, prepare 5 mL of sterile freezing media (90% FBS, 10% DMSO v/v) per stock plate.
- 6.
- After centrifugation use an 8- or 12-channel pipette to carefully remove supernatant from the pelleted cells in the stock plate.
CRITICAL STEP Take care not to dislodge and discard cells by disturbing the pellet.
- 7.
- Quickly and carefully resuspend cells in 50 µL of freezing buffer.
- 8.
- Wrap the stock plate in parafilm, place in polystyrene box or freezing box and store at −80 °C.
3.3. Cell Lysis. Time for Completion: Overnight (18 h)
- 9.
- Prepare 3 mL lysis buffer per full plate by adding proteinase K (see Section 5).
- 10.
- Pellet cells in the genotyping plate from step 4 in Section 3.2 (250× g, 5 min, room temperature).
- 11.
- Using an 8- or 12-channel pipette remove supernatant and resuspend in 30 µL lysis buffer.
- 12.
- Seal plate and incubate at 37 °C overnight.
- 13.
- On the following day, heat to 95 °C for 10 min in thermocycler to deactivate proteinase K.
- 14.
- Cool to 4 °C
 PAUSE STEP Lysed cells can be stored at −20 °C for up to one month or at −80 °C for one year.
PAUSE STEP Lysed cells can be stored at −20 °C for up to one month or at −80 °C for one year.3.4. Locus Amplification and PCR Product Clean-Up. Time for Completion: 4 h
- 15.
- On ice, prepare 1150 µL of Locus PCR master mix per lysed plate by combining 1125 µL Platinum master mix and 25 µL of 10 µM locus primer mix (Section 2.3).
- 16.
- Aliquot 11.5 µL of Locus PCR master mix into each well of a 96-well PCR plate on ice.
- 17.
- On ice, add 1 µL of cell lysate to the corresponding well of the PCR plate and mix by pipetting.
- 18.
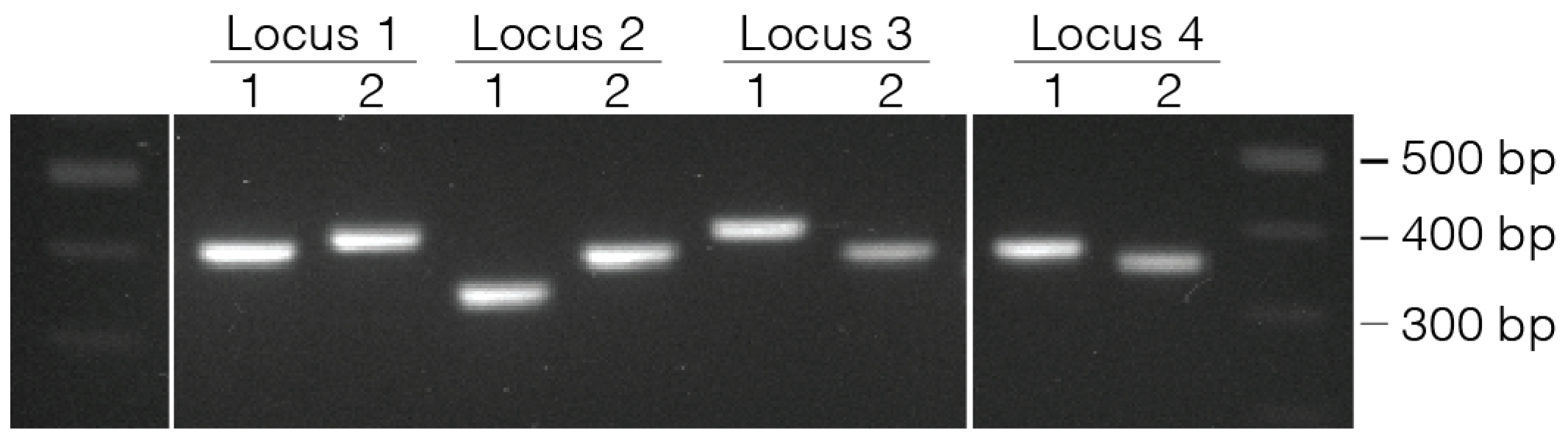
- Place in thermocycler and amplify using the Platinum PCR cycling settings in Table 2.
- 19.
- Transfer 1 µL of PCR products to the corresponding well of a new 96-well PCR plate.
- 20.
- Prepare 220 µL PCR clean-up master mix per plate by combining 16.5 µL exonuclease I, 16.5 µL shrimp alkaline phosphatase (SAP) and 187 µL PCR grade water.
- 21.
- Add 2 µL of PCR clean-up master mix to each well, mix by pipetting.
- 22.
- Incubate in a thermocycler at 37 °C for 30 min, 85 °C for 15 min, and then cool to 4 °C.
PAUSE STEP After stopping the reaction, the mix can be stored at 4 °C overnight.3.5. Well Barcoding and PCR Product Clean-Up. Time for Completion: 3 h
- 23.
- To each well of cleaned-up PCR product add 11.5 µL Platinum master mix (perform on ice).
- 24.
- Prepare a stock 96-well plate of barcoding primers by combining all unique pairs of row and column primers at 5 µM each by adding equal volumes (2–5 µL) of each iC7 and iR5 primer at 10 µM. This primer plate may be stored at −20 °C and used multiple times.
- 25.
- Using the barcoding primers prepared in step 24 and a multichannel pipette add 0.5 µL of primers to the appropriate wells of the genotyping plate.
- 26.
- Place in a thermocycler and amplify using the Platinum PCR cycling settings in Table 2.
- 27.
- During amplification bring an aliquot of 875 µL Ampure XP beads to room temp in an Eppendorf tube.
- 28.
- Pool 5 µL from each well of a single plate into a single 1.5 mL DNA low bind tube (480 µL in total). Store excess PCR reaction at −20 °C.
- 29.
- Add 864 µL Ampure XP beads (1.8 volumes) to the pooled PCR products. Mix by pipetting.
- 30.
- Incubate at room temp for 5 min.
- 31.
- During incubation prepare 80% ethanol (1200 µL 100% ethanol, 300 µL PCR grade water).
- 32.
- Place on magnetic stand. After the liquid has cleared ~5 min), remove and discard the liquid without disturbing the beads.
- 33.
- Add 700 µL of fresh 80% ethanol again without disturbing beads. Incubate 30 s.
- 34.
- Remove the ethanol and the repeat wash with another 700 µL of fresh 80% ethanol.
- 35.
- Discard the ethanol, spin briefly on a bench-top centrifuge and replace on magnetic stand.
- 36.
- Discard the residual ethanol and allow to air dry until beads appear matte in appearance ~5 min).
CRITICAL STEP Do not over dry the beads as this will reduce yield. Beads should appear like damp mud, neither glossy wet nor dry. Cracks in the bead pellet are indicative of over-drying.
- 37.
- Remove tube from magnet and resuspend beads in 55 µL of PCR grade water by pipetting 10 times.
- 38.
- Incubate at room temp for 2 min.
- 39.
- Replace on magnetic stand.
- 40.
- Once clear (~4 min) recover 53 µL of eluted PCR product and transfer to a new 1.5 mL DNA low-bind tube.
- 41.
- Use 2 µL of eluted PCR to quantify the DNA concentration using a Qubit BR DNA kit.
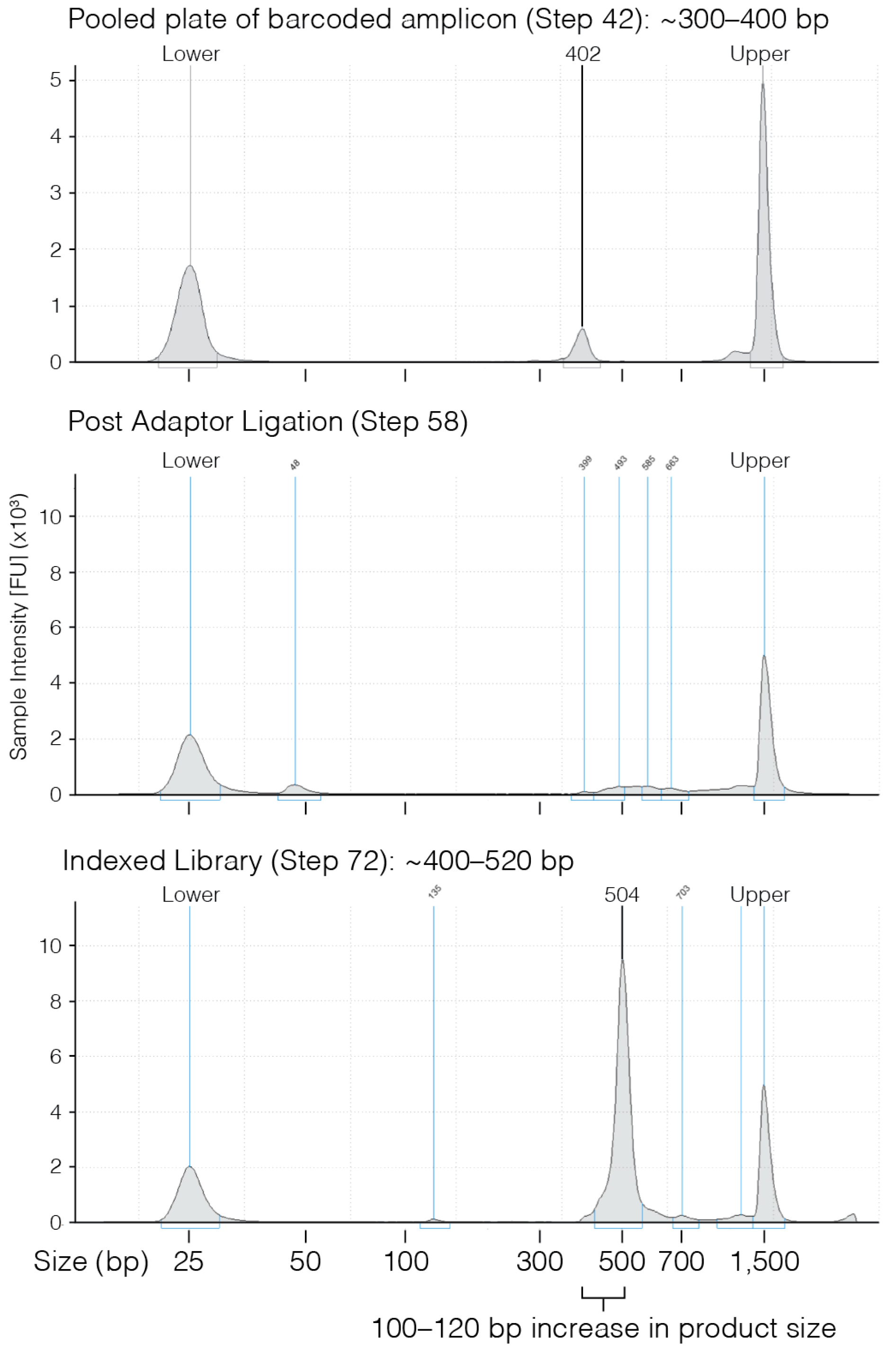
- 42.
- Use 1 µL of eluted PCR to evaluate the product size on a D1000 TapeStation gel.
PAUSE STEP After purification the products can be stored at 4 °C overnight or at −20 °C for several months.3.6. Addition of Illumina Adaptors. Time for Completion: 3 h
- 43.
- Combine 50 µL of DNA (0.5–2 µg), 7 µL 10× End Prep Buffer and 3 µL End Prep Enzyme. Mix by pipetting and incubate at 20 °C for 30 min in a thermocycler (lid open or un-heated).
- 44.
- Increase temperature to 65 °C and incubate for 30 min (lid heated to 75 °C).
- 45.
- Add 30 µL Ultra II Ligation Master Mix, 2.5 µL NEBNext Adaptor, 1 µL Ligation enhancer. Mix by pipetting and incubate at 20 °C for 15 min.
- 46.
- Add 3 µL USERTM enzyme. Mix by pipetting and incubate at 37 °C for 15 min.
- 47.
- During this USERTM enzyme reaction warm 90 µL of Ampure XP beads to room temp.
- 48.
- Add 87 µL Ampure XP beads (0.9×) and mix by pipetting. Incubate at room temp for 5 min.
- 49.
- During incubation prepare 80% ethanol (800 µL 100% ethanol, 200 µL PCR grade water).
- 50.
- Place on magnetic stand. Without disturbing beads, discard liquid when clear (~5 min).
- 51.
- Add 500 µL of fresh 80% ethanol without disturbing beads. Incubate 30 s.
- 52.
- Remove ethanol and repeat wash with another 500 µL of fresh 80% ethanol.
- 53.
- Discard ethanol, spin briefly on a bench-top centrifuge and replace on magnetic stand.
- 54.
- Discard residual ethanol and allow to air dry until beads are matte in appearance (~5 min).
CRITICAL STEP Do not over dry the beads as this will reduce yield. Beads should appear like damp mud, neither glossy wet nor dry. Cracks in the bead pellet are indicative of over-drying.
- 55.
- Remove tube from magnet and resuspend beads in 30.5 µL of PCR grade water by pipetting 10 times.
- 56.
- Incubate at room temperature for 2 min.
- 57.
- Replace on magnetic stand.
- 58.
- Once clear ~4 min) recover 28.5 µL of eluted PCR product and transfer to a new PCR tube.
3.7. Indexing for Multiplexing of Multiple Plates. Time for Completion: 2 h
- 59.
- To the adaptor ligated library add:
- -
- 5 µL NEB Universal Primer
- -
- 5 µL NEB Index Primer
- -
- 10 µL Herculase II 5× Buffer
- -
- 0.5 µL deoxynucleotide triphosphate (dNTP)
- -
- 1 µL Herculase II Polymerase
- 60.
- Mix by pipetting and amplify using the Herculase PCR cycling settings in Table 3.
- 61.
- During the PCR warm 40 µL of Ampure XP beads to room temp.
- 62.
- Add 40 µL Ampure XP beads (0.8×) to the PCR and mix by pipetting. Incubate at room temperature for 5 min.
- 63.
- During incubation prepare 80% ethanol (800 µL 100% ethanol, 200 µL PCR grade water).
- 64.
- Place on magnetic stand. Without disturbing beads, discard liquid when clear ~5 min).
- 65.
- Add 500 µL of fresh 80% ethanol without disturbing beads. Incubate 30 s.
- 66.
- Remove ethanol and repeat wash with another 500 µL of fresh 80% ethanol.
- 67.
- Discard ethanol, spin briefly on a bench-top centrifuge and replace on magnetic stand.
- 68.
- Discard residual ethanol and allow to air dry until beads are matte in appearance (~5 min).
CRITICAL STEP Do not over dry the beads as this will reduce yield. Beads should appear like damp mud, neither glossy wet nor dry. Cracks in the bead pellet are indicative of over-drying.
- 69.
- Remove tube from magnet and resuspend beads in 30 µL of 0.1× Tris-EDTA (TE) by pipetting 10 times.
- 70.
- Incubate at room temperature for 2 min.
- 71.
- Replace on magnetic stand. Once clear (~4 min) recover 28 µL of eluted PCR product and transfer to a new DNA low bind tube.
- 72.
- Confirm PCR product size using either a 2% agarose gel or D1000 TapeStation.
PAUSE STEP After this step the indexed library can be stored at 4 °C overnight or at −20 °C for several months.3.8. Quantification and Sequencing. Time for Completion: 28 h
- 73.
- Make a 1:10,000 and 1:20,000 dilution of indexed PCR products and quantify by real-time qPCR using the KAPA Illumina quantification kit.
- 74.
- If sequencing in-house prepare 8 nM dilutions of each PCR plate pool and combine differently indexed plates in equal volumes.
- 75.
- If sequencing in a core facility prepare DNA pools to their specifications.
- 76.
- Sequence on a MiSeq using a 500-cycle Nano kit (250 bp reads, paired end) with 10% PhiX.
CRITICAL STEP Locus-specific PCR products will have near identical sequences, PhiX is essential to avoid MiSeq run failure from low complexity. If sequencing a single locus specific PCR product, increase PhiX to 30%.3.9. Data Analysis. Time for Completion: 1–3 Days
- 77.
- Download fastq files from either BaseSpace or core facility provider.
- 78.
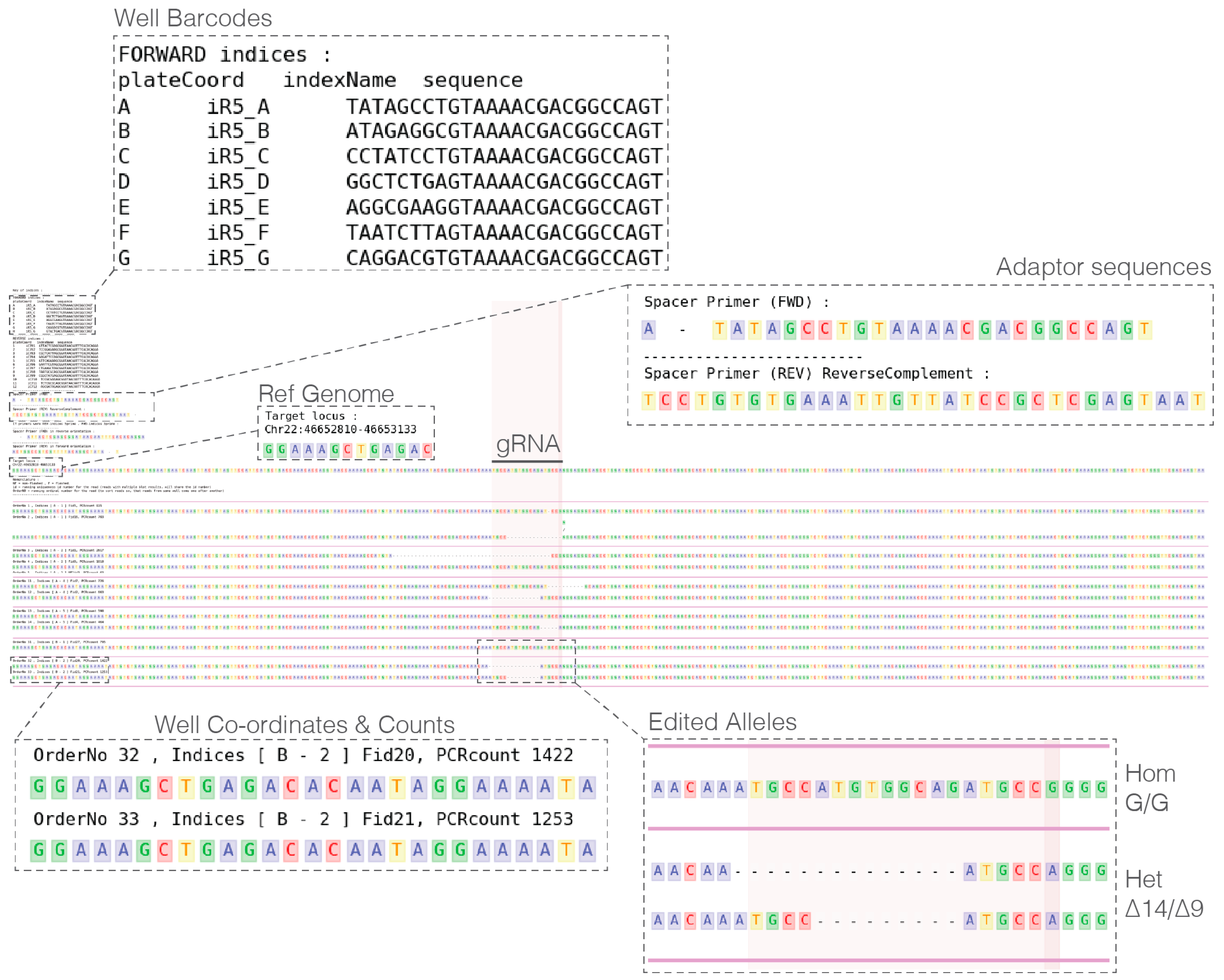
- In a single UNIX directory prepare the following metadata plain text files:
- PIPE_fastqPasths.txt: Containing names and paths to fastq files.
- PIPE_spacerBarcodePrimer_FWD.txt: Containing forward primer sequences.
- PIPE_spacerBarcodePrimer_REV.txt: Containing reverse primer sequences.
- PIPE_targetLocus_xxX.bed: Locus PCR amplification coordinates and additional region(s) of interest highlighted (Note: species of interest replaces xxX in the file name, e.g., PIPE_targetLocus_hg19.bed)
CRITICAL STEP Plain text files are essential as rich text files will not be read correctly and cause analysis failure.
- 79.
- Run analysis with the following minimal command (additional parameters are available): > ./plateScreen96.sh-g hg19.
- 80.
- Analyze pipe output and select clones to recover (see Expected Results).
3.10. Recovery of Clones of Interest. Time for Completion: 2 h
- 81.
- Prepare one 1.5 mL microcentrifuge tube per clone by adding 1 mL sterile phosphate-buffered saline (PBS).
- 82.
- Recover plate from −80 °C storage and remove parafilm.
- 83.
- Working quickly, add 150 µL of sterile PBS to wells containing clones to be recovered.
- 84.
- As each clone thaws, pipette mix to quickly combine with PBS, transfer melted clones to microcentrifuge tubes containing PBS.
- 85.
- Centrifuge to pellet cells (250× g, 5 min).
- 86.
- Discard supernatant and resuspend in appropriate amount of growth media and plate for expansion.
4. Expected Results
5. Reagents Setup
Cell Lysis Buffer
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Bradley, A. Repair of CRISPR–Cas9-induced double-stranded breaks leads to large deletions and complex rearrangements. Nature 2018. [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhang, Y.; Yao, S.; Wei, Y. A PCR Based Protocol for Detecting Indel Mutations Induced by TALENs and CRISPR/Cas9 in Zebrafish. PLoS ONE 2014, 9, e98282. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Yan, T.; Cheung, A.M.S.; Chuah, C.T.H.; Li, S. High-throughput genotyping of CRISPR/Cas9-mediated mutants using fluorescent PCR-capillary gel electrophoresis. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Samarut, É.; Lissouba, A.; Drapeau, P. A simplified method for identifying early CRISPR-induced indels in zebrafish embryos using High Resolution Melting analysis. BMC Genom. 2016, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, J.L.; Byrne, S.; Pan, J.; Church, G.M. CRISPR/Cas9-Directed Genome Editing of Cultured Cells. Curr. Protocols Mol. Biol. 2014, 107, 31.1.1–31.1.17. [Google Scholar]
- Schmid-Burgk, J.L.; Schmidt, T.; Gaidt, M.M.; Pelka, K.; Latz, E.; Ebert, T.S.; Hornung, V. OutKnocker: A web tool for rapid and simple genotyping of designer nuclease edited cell lines OutKnocker: A web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Moir-Meyer, G.; Cheong, P.L.; Olijnik, A.; Brown, J.; Knight, S.; King, A.; Kurita, R.; Nakamura, Y.; Gibbons, R.; Higgs, D.; et al. Robust CRISPR/Cas9 Genome Editing of the HUDEP-2 Erythroid Precursor Line Using Plasmids and Single-Stranded Oligonucleotide Donors. Methods Protoc. 2018, 1, 28. [Google Scholar] [CrossRef]
- Telenius, J.M. plateScreen96 (GitHub). Available online: github.com/Hughes-Genome-Group/plateScreen96/releases (accessed on 26 July 2018).
- Telenius, J.M. plate96 Guide. plateSreen96—CRISPR short indel genotype analyser. Available online: http://sara.molbiol.ox.ac.uk/public/telenius/plate96 (accessed on 26 July 2018).
- Integrated DNA Technologies (IDT). PrimerQuest Tool. Available online: www.idtdna.com/Primerquest (accessed on 26 July 2018).
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Barcode | Sequence |
|---|---|---|
| iR5 – Row Barcode Primers (Forward) | ||
| iR5_A | TATAGCCT | GATTATAGCCTGTAAAACGACGGCCAGT |
| iR5_B | ATAGAGGC | GATATAGAGGCGTAAAACGACGGCCAGT |
| iR5_C | CCTATCCT | GATCCTATCCTGTAAAACGACGGCCAGT |
| iR5_D | GGCTCTGA | GATGGCTCTGAGTAAAACGACGGCCAGT |
| iR5_E | AGGCGAAG | GATAGGCGAAGGTAAAACGACGGCCAGT |
| iR5_F | TAATCTTA | GATTAATCTTAGTAAAACGACGGCCAGT |
| iR5_G | CAGGACGT | GATCAGGACGTGTAAAACGACGGCCAGT |
| iR5_H | GTACTGAC | GATGTACTGACGTAAAACGACGGCCAGT |
| iC7 – Column Barcode Primers (Reverse) | ||
| iC701 | ATTACTCG | GATATTACTCGAGCGGATAACAATTTCACACAGGA |
| iC702 | TCCGGAGA | GATTCCGGAGAAGCGGATAACAATTTCACACAGGA |
| iC703 | CGCTCATT | GATCGCTCATTAGCGGATAACAATTTCACACAGGA |
| iC704 | GAGATTCC | GATGAGATTCCAGCGGATAACAATTTCACACAGGA |
| iC705 | ATTCAGAA | GATATTCAGAAAGCGGATAACAATTTCACACAGGA |
| iC706 | GAATTCGT | GATGAATTCGTAGCGGATAACAATTTCACACAGGA |
| iC707 | CTGAAGCT | GATCTGAAGCTAGCGGATAACAATTTCACACAGGA |
| iC708 | TAATGCGC | GATTAATGCGCAGCGGATAACAATTTCACACAGGA |
| iC709 | CGGCTATG | GATCGGCTATGAGCGGATAACAATTTCACACAGGA |
| iC710 | TCCGCGAA | GATTCCGCGAAAGCGGATAACAATTTCACACAGGA |
| iC711 | TCTCGCGC | GATTCTCGCGCAGCGGATAACAATTTCACACAGGA |
| iC712 | AGCGATAG | GATAGCGATAGAGCGGATAACAATTTCACACAGGA |
| Step | Temp. | Time |
|---|---|---|
| Step 1 | 94 °C | 2 min |
| Step 2 | 94 °C | 30 s |
| Step 3 | 62 °C | 30 s |
| Step 4 | 68 °C | 1 min |
| Repeat steps 2–4 for a total of 12 cycles | ||
| Step 5 | 72 °C | 10 min |
| Step 6 | 4 °C | ∞ |
| Step | Temp. | Time |
|---|---|---|
| Step 1 | 98 °C | 30 s |
| Step 2 | 98 °C | 10 s |
| Step 3 | 65 °C | 30 s |
| Step 4 | 72 °C | 30 s |
| Repeat steps 2–4 for a total of 6 cycles | ||
| Step 5 | 72 °C | 5 min |
| Step 6 | 4 °C | ∞ |
| Wells Processed | Successfully Sequenced | Success Rate | |
|---|---|---|---|
| Locus 1 | 52 | 49 | 94.2% |
| Locus 2 | 244 | 217 | 88.9% |
| Locus 3 | 87 | 85 | 97.7% |
| Locus 4 | 59 | 53 | 89.8% |
| Locus 5 | 47 | 43 | 91.4% |
| Locus 6 | 74 | 72 | 97.2% |
| Locus 7 | 236 | 234 | 99.1% |
| Total | 799 | 753 | 94.2% |
| Reagent | Volume | Final Conc. |
|---|---|---|
| 1 M Tris, pH 8.0 | 150 µL | 50 mM |
| 0.5 M EDTA, pH 8.0 | 6 µL | 1 mM |
| 50% Tween 20 | 30 µL | 0.5% |
| PCR Grade Water | 2.805 mL | - |
| Proteinase K (>0.6 U/µL) | 9 µL | >0.0018 U/µL |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nussbaum, L.; Telenius, J.M.; Hill, S.; Hirschfeld, P.P.; Suciu, M.C.; The WIGWAM Consortium; Downes, D.J.; Hughes, J.R. High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates. Methods Protoc. 2018, 1, 29. https://doi.org/10.3390/mps1030029
Nussbaum L, Telenius JM, Hill S, Hirschfeld PP, Suciu MC, The WIGWAM Consortium, Downes DJ, Hughes JR. High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates. Methods and Protocols. 2018; 1(3):29. https://doi.org/10.3390/mps1030029
Chicago/Turabian StyleNussbaum, Lea, Jelena M. Telenius, Stephanie Hill, Priscila P. Hirschfeld, Maria C. Suciu, The WIGWAM Consortium, Damien J. Downes, and Jim R. Hughes. 2018. "High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates" Methods and Protocols 1, no. 3: 29. https://doi.org/10.3390/mps1030029
APA StyleNussbaum, L., Telenius, J. M., Hill, S., Hirschfeld, P. P., Suciu, M. C., The WIGWAM Consortium, Downes, D. J., & Hughes, J. R. (2018). High-Throughput Genotyping of CRISPR/Cas Edited Cells in 96-Well Plates. Methods and Protocols, 1(3), 29. https://doi.org/10.3390/mps1030029