
Whole-Genome Sequencing Can Identify Clinically Relevant Variants from a Single Sub-Punch of a Dried Blood Spot Specimen
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.1.1. Consent and Ethics Approval
2.1.2. Dried Blood Spot Collection
2.1.3. DNA Extraction from Venous Blood
2.2. DNA Quantification
2.3. NGS Library Preparation
2.4. Sequencing Library Quantification, Pooling, and Sequencing
2.5. Data Analysis
3. Results
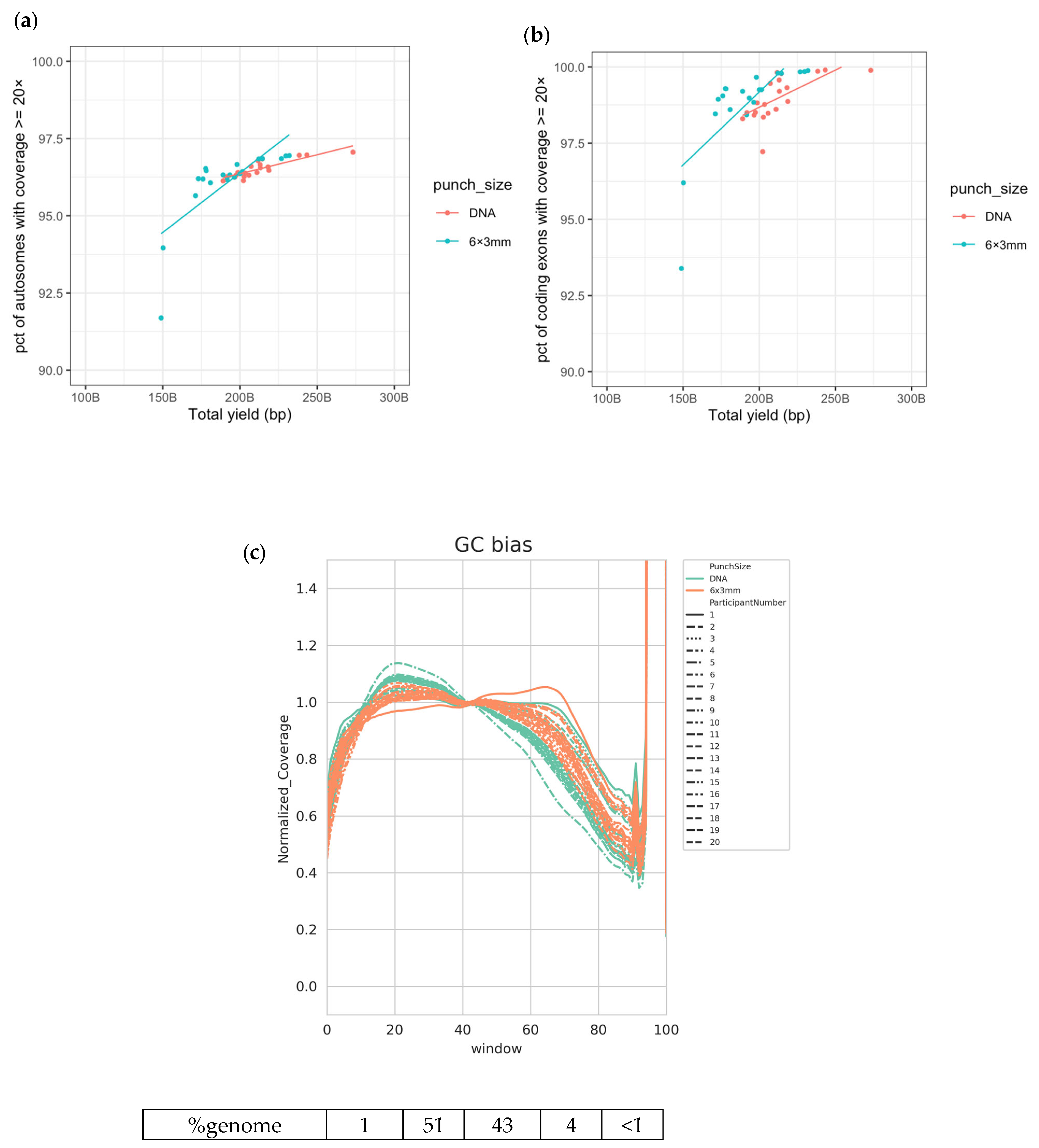
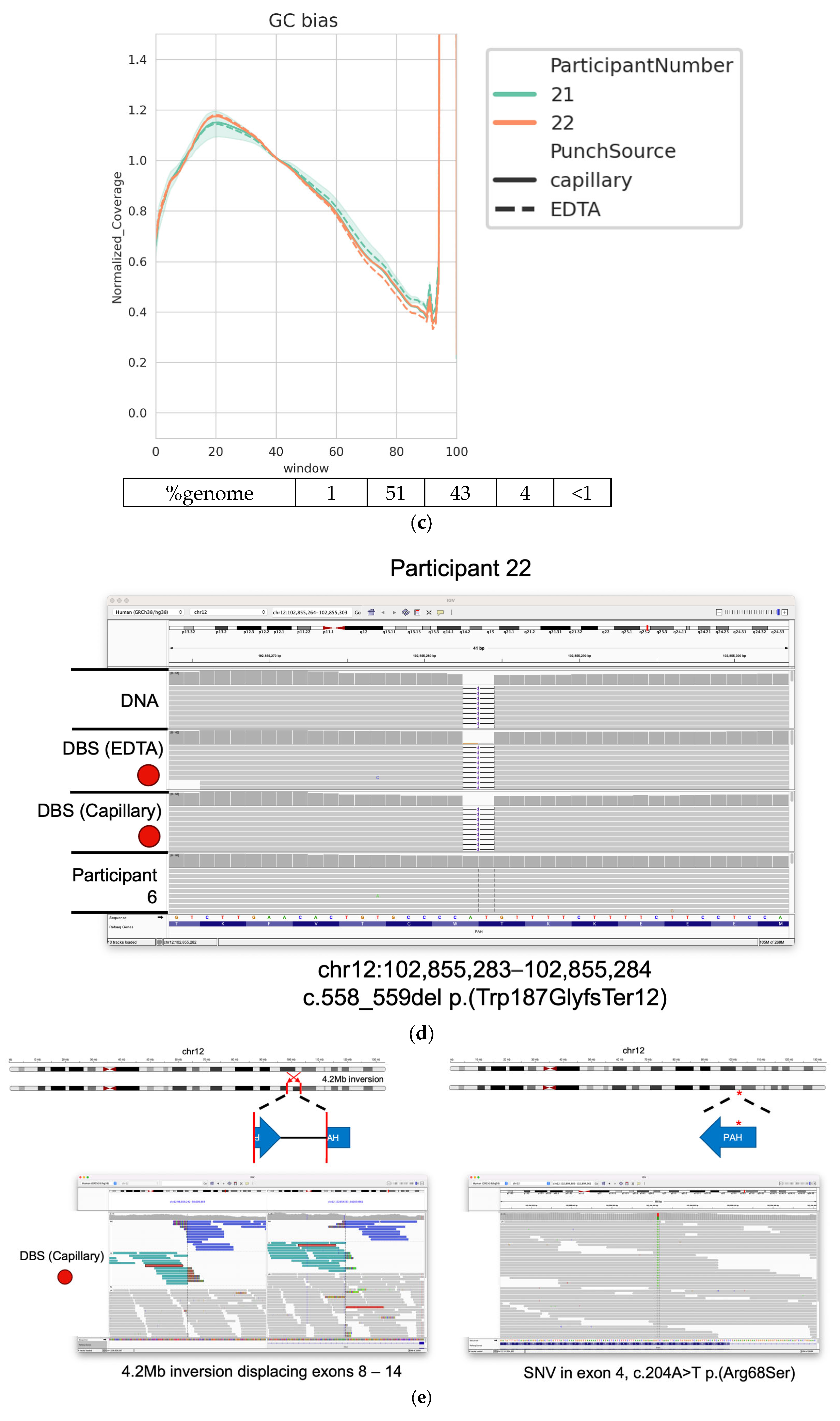
3.1. High-Quality WGS Data Are Achieved from DBS
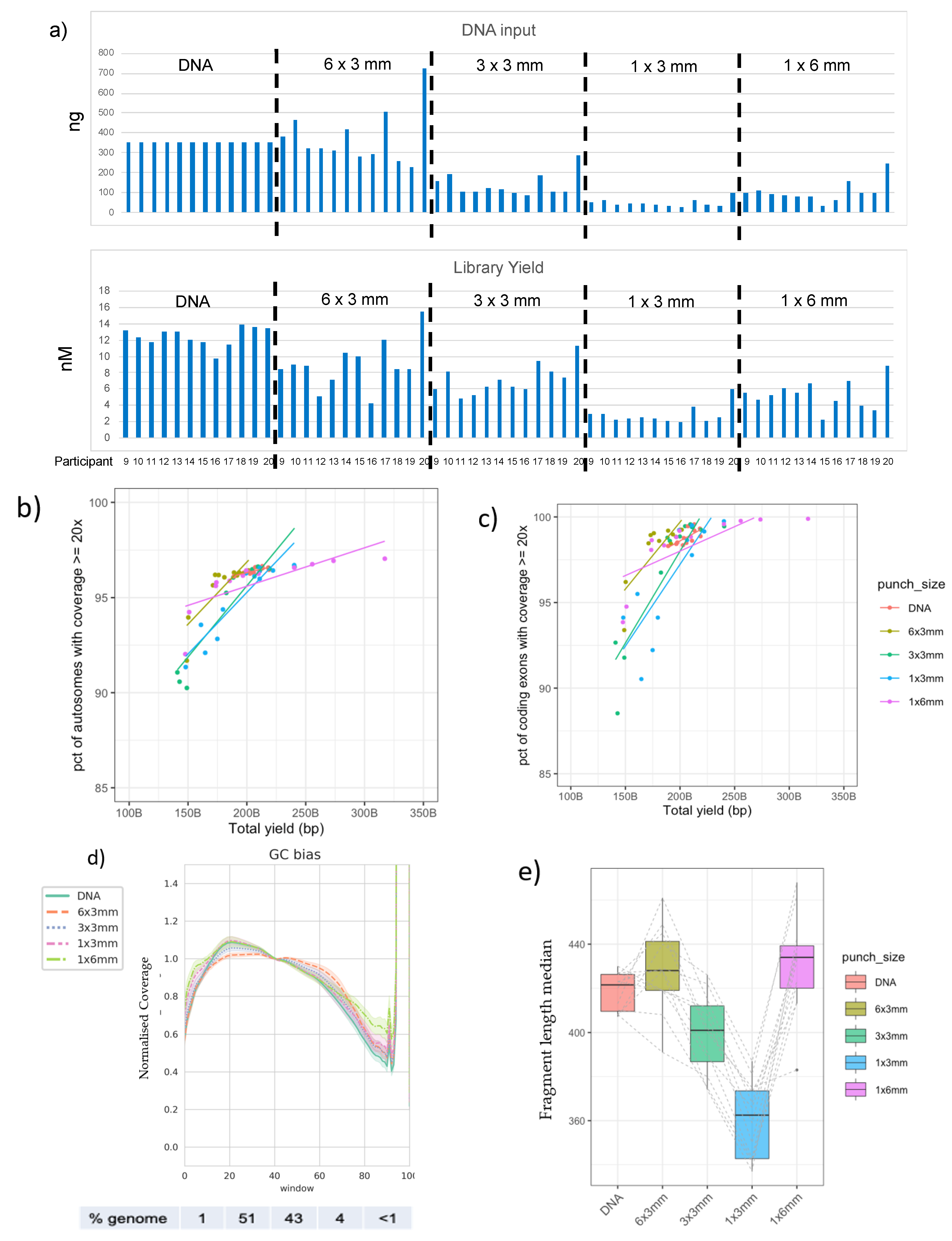
3.2. Good-Quality WGS Data Can Be Achieved from a Variety of DBS Punch Configurations
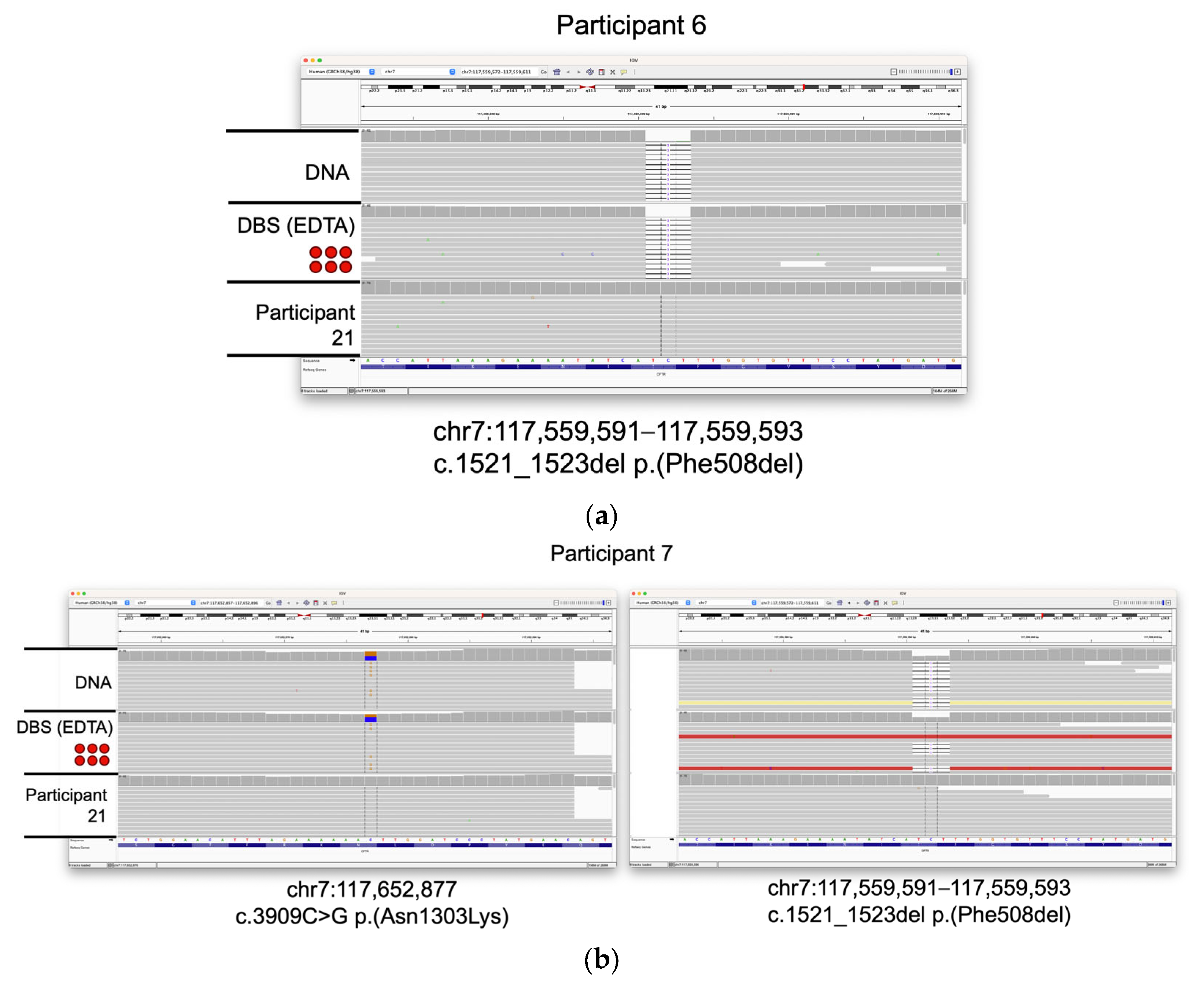
3.3. Identification of Clincally Relevant Variants from DBSs
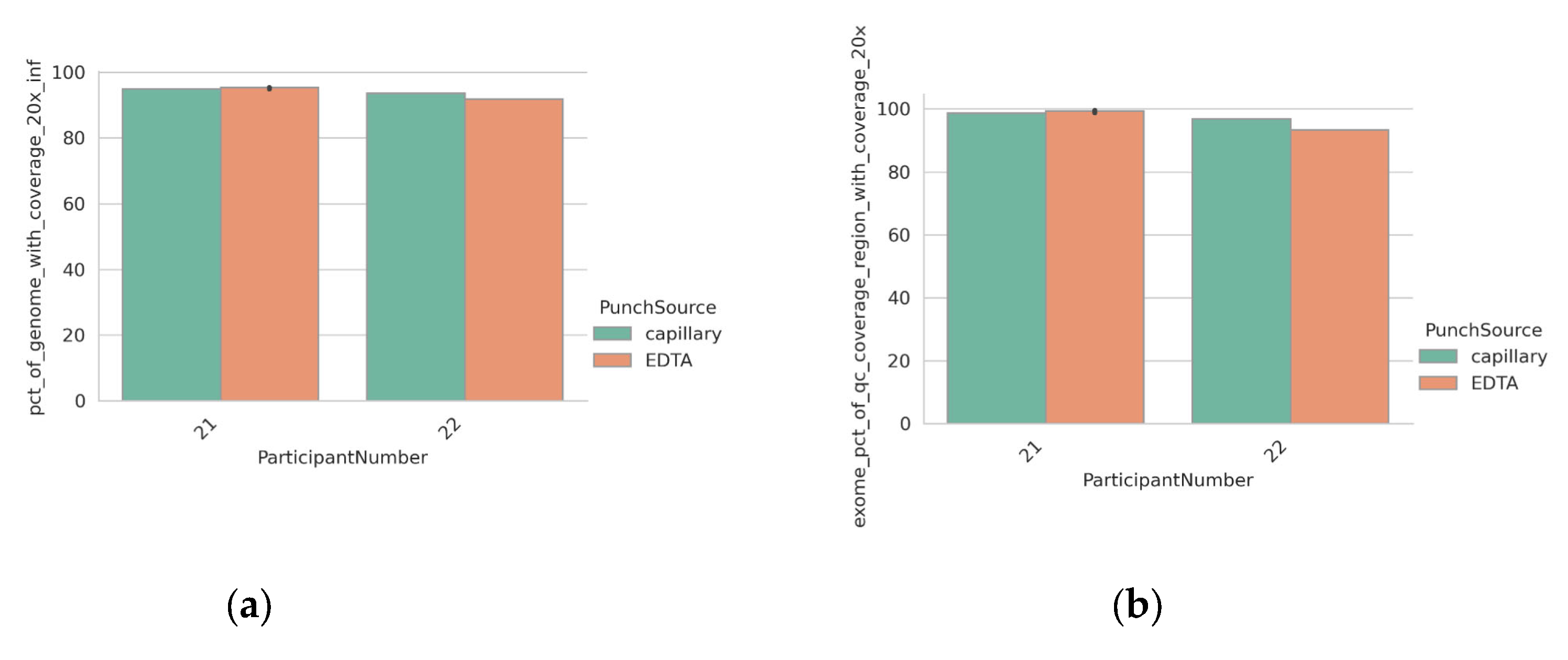
3.4. Detection of Clinically Relevant Variants from DBS Taken Directly from the Participant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Anesth. Analg. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Deignan, J.L.; Astbury, C.; Cutting, G.R.; del Gaudio, D.; Gregg, A.R.; Grody, W.W.; Monaghan, K.G.; Richards, S. CFTR variant testing: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Anesth. Analg. 2020, 22, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- The 100,000 Genomes Project Pilot Investigators. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report. N. Engl. J. Med. 2021, 385, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Ajay, S.S.; Rajan, V.; Brown, C.; Bluske, K.; Burns, N.J.; Chawla, A.; Coffey, A.J.; Malhotra, A.; Scocchia, A.; et al. Copy-number variants in clinical genome sequencing: Deployment and interpretation for rare and undiagnosed disease. Anesth. Analg. 2018, 21, 1121–1130. [Google Scholar] [CrossRef]
- Whitford, W.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Evaluation of the performance of copy number variant prediction tools for the detection of deletions from whole genome sequencing data. J. Biomed. Inform. 2019, 94, 103174. [Google Scholar] [CrossRef] [PubMed]
- Austin-Tse, C.A.; Jobanputra, V.; Perry, D.L.; Bick, D.; Taft, R.J.; Venner, E.; Gibbs, R.A.; Young, T.; Barnett, S.; Belmont, J.W.; et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. npj Genom. Med. 2022, 7, 27. [Google Scholar] [CrossRef]
- Liu, Z.; Roberts, R.; Mercer, T.R.; Xu, J.; Sedlazeck, F.J.; Tong, W. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. 2022, 23, 68. [Google Scholar] [CrossRef]
- Coutelier, M.; Holtgrewe, M.; Jäger, M.; Flöttman, R.; Mensah, M.A.; Spielmann, M.; Krawitz, P.; Horn, D.; Beule, D.; Mundlos, S. Combining callers improves the detection of copy number variants from whole-genome sequencing. Eur. J. Hum. Genet. 2022, 30, 178–186. [Google Scholar] [CrossRef]
- Dolzhenko, E.; Bennett, M.F.; Richmond, P.A.; Trost, B.; Chen, S.; van Vugt, J.J.F.A.; Nguyen, C.; Narzisi, G.; Gainullin, V.G.; Gross, A.M.; et al. ExpansionHunter Denovo: A computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020, 21, 102. [Google Scholar] [CrossRef]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Abu Alnadi, N.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid Whole-Genome Sequencing for Genetic Disease Diagnosis in Neonatal Intensive Care Units. Sci. Transl. Med. 2012, 4, 154ra135. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D.; Medne, L.; Weatherly, J.M.; Wild, K.T.; Biswas, S.; Devkota, B.; Hartman, T.; Brunelli, L.; Fishler, K.P.; Abdul-Rahman, O.; et al. Effect of Whole-Genome Sequencing on the Clinical Management of Acutely Ill Infants with Suspected Genetic Disease: A Randomized Clinical Trial. JAMA Pediatr. 2021, 175, 1218–1226. [Google Scholar] [PubMed]
- Clark, M.M.; Hildreth, A.; Batalov, S.; Ding, Y.; Chowdhury, S.; Watkins, K.; Ellsworth, K.; Camp, B.; Kint, C.I.; Yacoubian, C.; et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019, 11, aat6177. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Cakici, J.A.; Clark, M.M.; Gaughran, M.; Feddock, M.; Batalov, S.; Bainbridge, M.N.; Carroll, J.; Caylor, S.A.; Clarke, C.; et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am. J. Hum. Genet. 2019, 105, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Lagerstedt-Robinson, K.; Magnusson, M.; Kvarnung, M.; Nilsson, D.; Lesko, N.; Engvall, M.; Anderlid, B.-M.; Arnell, H.; Johansson, C.B.; et al. Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Bowdin, S.; Hayeems, R.; Monfared, N.; Cohn, R.; Meyn, M. The SickKids Genome Clinic: Developing and evaluating a pediatric model for individualized genomic medicine. Clin. Genet. 2016, 89, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Willmen, T.; Willmen, L.; Pankow, A.; Ronicke, S.; Gabriel, H.; Wagner, A.D. Rare diseases: Why is a rapid referral to an expert center so important? BMC Health Serv. Res. 2023, 23, 904. [Google Scholar] [CrossRef]
- Bauskis, A.; Strange, C.; Molster, C.; Fisher, C. The diagnostic odyssey: Insights from parents of children living with an undiagnosed condition. Orphanet J. Rare Dis. 2022, 17, 233. [Google Scholar] [CrossRef]
- Farnaes, L.; Hildreth, A.; Sweeney, N.M.; Clark, M.M.; Chowdhury, S.; Nahas, S.; Cakici, J.A.; Benson, W.; Kaplan, R.H.; Kronick, R.; et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. npj Genom. Med. 2018, 3, 10. [Google Scholar] [CrossRef]
- Splinter, K.; Adams, D.R.; Bacino, C.A.; Bellen, H.J.; Bernstein, J.A.; Cheatle-Jarvela, A.M.; Eng, C.M.; Esteves, C.; Gahl, W.A.; Hamid, R.; et al. Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N. Engl. J. Med. 2018, 379, 2131–2139. [Google Scholar] [CrossRef]
- Petrikin, J.E.; Cakici, J.A.; Clark, M.M.; Willig, L.K.; Sweeney, N.M.; Farrow, E.G.; Saunders, C.J.; Thiffault, I.; Miller, N.A.; Zellmer, L.; et al. The NSIGHT1-randomized controlled trial: Rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. npj Genom. Med. 2018, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Scocchia, A.; Wigby, K.M.; Masser-Frye, D.; Del Campo, M.; Galarreta, C.I.; Thorpe, E.; McEachern, J.; Robinson, K.; Gross, A.; ICSL Interpretation and Reporting Team; et al. Clinical whole genome sequencing as a first-tier test at a resource-limited dysmorphology clinic in Mexico. npj Genom. Med. 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Consortium, T.B. Dispatches from Biotech beginning BeginNGS: Rapid newborn genome sequencing to end the diagnostic and therapeutic odyssey. Am. J. Med. Genet. Part C Semin. Med. Genet. 2022, 190, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Kingsmore, S.F.; Smith, L.D.; Kunard, C.M.; Bainbridge, M.; Batalov, S.; Benson, W.; Blincow, E.; Caylor, S.; Chambers, C.; Del Angel, G.; et al. A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. Am. J. Hum. Genet. 2022, 109, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B., Jr.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef] [PubMed]
- Seydel, C. Baby’s first genome. Nat. Biotechnol. 2022, 40, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, D.; Caylor, S.; Waldman, B.; Benson, W.; Ashburner, C.; Carmichael, J.L.; Carroll, J.; Cham, E.; Chowdhury, S.; Cleary, J.; et al. Project Baby Bear: Rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am. J. Hum. Genet. 2021, 108, 1231–1238. [Google Scholar] [CrossRef]
- Mestek-Boukhibar, L.; Clement, E.; Jones, W.D.; Drury, S.; Ocaka, L.; Gagunashvili, A.; Stabej, P.L.Q.; Bacchelli, C.; Jani, N.; Rahman, S.; et al. Rapid Paediatric Sequencing (RaPS): Comprehensive real-life workflow for rapid diagnosis of critically ill children. J. Med. Genet. 2018, 55, 721–728. [Google Scholar] [CrossRef]
- Ding, Y.; Owen, M.; Le, J.; Batalov, S.; Chau, K.; Kwon, Y.H.; Van Der Kraan, L.; Bezares-Orin, Z.; Zhu, Z.; Veeraraghavan, N.; et al. Scalable, high quality, whole genome sequencing from archived, newborn, dried blood spots. npj Genom. Med. 2023, 8, 5. [Google Scholar] [CrossRef]
- Agrawal, P.; Katragadda, S.; Hariharan, A.K.; Raghavendrachar, V.G.; Agarwal, A.; Dayalu, R.; Awasthy, D.; Sharma, S.C.; Sivasamy, Y.K.; Lakshmana, P.; et al. Validation of whole genome sequencing from dried blood spots. BMC Med. Genom. 2021, 14, 110. [Google Scholar] [CrossRef]
- Poulsen, J.B.; Lescai, F.; Grove, J.; Bækvad-Hansen, M.; Christiansen, M.; Hagen, C.M.; Maller, J.; Stevens, C.; Li, S.; Li, Q.; et al. High-Quality Exome Sequencing of Whole-Genome Amplified Neonatal Dried Blood Spot DNA. PLoS ONE 2016, 11, e0153253. [Google Scholar] [CrossRef] [PubMed]
- Hollegaard, M.V.; Grauholm, J.; Nielsen, R.; Grove, J.; Mandrup, S.; Hougaard, D.M. Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. Mol. Genet. Metab. 2013, 110, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bassaganyas, L.; Freedman, G.; Vaka, D.; Wan, E.; Lao, R.; Chen, F.; Kvale, M.; Currier, R.J.; Puck, J.M.; Kwok, P.-Y. Whole exome and whole genome sequencing with dried blood spot DNA without whole genome amplification. Hum. Mutat. 2018, 39, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Roman, T.S.; Crowley, S.B.; Roche, M.I.; Foreman, A.K.M.; O’daniel, J.M.; Seifert, B.A.; Lee, K.; Brandt, A.; Gustafson, C.; DeCristo, D.M.; et al. Genomic Sequencing for Newborn Screening: Results of the NC NEXUS Project. Am. J. Hum. Genet. 2020, 107, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Grange, D.K.; Milanowski, A.; Vockley, G.; Feillet, F.; Crombez, E.A.; Abadie, V.; Harding, C.O.; Cederbaum, S.; Dobbelaere, D.; et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): A phase II, multicentre, open-label, screening study. J. Inherit. Metab. Dis. 2007, 30, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Ajay, S.S.; Parker, S.C.; Abaan, H.O.; Fajardo, K.V.F.; Margulies, E.H. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011, 21, 1498–1505. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Ding, L.; Mardis, E.R.; Wilson, R.K. Challenges of sequencing human genomes. Brief. Bioinform. 2010, 11, 484–498. [Google Scholar] [CrossRef]
- Moat, S.J.; George, R.S.; Carling, R.S. Use of Dried Blood Spot Specimens to Monitor Patients with Inherited Metabolic Disorders. Int. J. Neonatal Screen. 2020, 6, 26. [Google Scholar] [CrossRef]
- Moat, S.J.; Zelek, W.M.; Carne, E.; Ponsford, M.J.; Bramhall, K.; Jones, S.; El-Shanawany, T.; Wise, M.P.; Thomas, A.; George, C.; et al. Development of a high-throughput SARS-CoV-2 antibody testing pathway using dried blood spot specimens. Ann. Clin. Biochem. 2021, 58, 123–131. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Turner, D.; Mesirov, J.P. igv.js: An embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 2022, 39, btac830. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Wu, Y.; Narzisi, G.; ORawe, J.A.; Barrón, L.T.; Rosenbaum, J.; Ronemus, M.; Iossifov, I.; Schatz, M.C.; Lyon, G.J. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Soper, E.R.; Braganza, G.T.; Rodriguez, J.E.; Zeid, N.; Cullina, S.; Bobo, D.; Moscati, A.; Merkelson, A.; Loos, R.J.F.; et al. Implementing genomic screening in diverse populations. Genome Med. 2021, 13, 17. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Team, C.G.; Soper, E.; Odgis, J.A.; Cullina, S.; Bobo, D.; Moscati, A.; Rodriguez, J.E.; Loos, R.; Cho, J.H.; et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019, 12, 2. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019, 12, 3. [Google Scholar] [CrossRef]
- Rehm, H.L.; Fowler, D.M. Keeping up with the genomes: Scaling genomic variant interpretation. Genome Med. 2019, 12, 5. [Google Scholar] [CrossRef]
- Gao, H.; Hamp, T.; Ede, J.; Schraiber, J.G.; McRae, J.; Singer-Berk, M.; Yang, Y.; Dietrich, A.S.D.; Fiziev, P.P.; Kuderna, L.F.K.; et al. The landscape of tolerated genetic variation in humans and primates. Science 2023, 380, eabn8153. [Google Scholar] [CrossRef]
- Fiziev, P.P.; McRae, J.; Ulirsch, J.C.; Dron, J.S.; Hamp, T.; Yang, Y.; Wainschtein, P.; Ni, Z.; Schraiber, J.G.; Gao, H.; et al. Rare penetrant mutations confer severe risk of common diseases. Science 2023, 380, eabo1131. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McBride, D.J.; Fielding, C.; Newington, T.; Vatsiou, A.; Fischl, H.; Bajracharya, M.; Thomson, V.S.; Fraser, L.J.; Fujita, P.A.; Becq, J.; et al. Whole-Genome Sequencing Can Identify Clinically Relevant Variants from a Single Sub-Punch of a Dried Blood Spot Specimen. Int. J. Neonatal Screen. 2023, 9, 52. https://doi.org/10.3390/ijns9030052
McBride DJ, Fielding C, Newington T, Vatsiou A, Fischl H, Bajracharya M, Thomson VS, Fraser LJ, Fujita PA, Becq J, et al. Whole-Genome Sequencing Can Identify Clinically Relevant Variants from a Single Sub-Punch of a Dried Blood Spot Specimen. International Journal of Neonatal Screening. 2023; 9(3):52. https://doi.org/10.3390/ijns9030052
Chicago/Turabian StyleMcBride, David J., Claire Fielding, Taksina Newington, Alexandra Vatsiou, Harry Fischl, Maya Bajracharya, Vicki S. Thomson, Louise J. Fraser, Pauline A. Fujita, Jennifer Becq, and et al. 2023. "Whole-Genome Sequencing Can Identify Clinically Relevant Variants from a Single Sub-Punch of a Dried Blood Spot Specimen" International Journal of Neonatal Screening 9, no. 3: 52. https://doi.org/10.3390/ijns9030052
APA StyleMcBride, D. J., Fielding, C., Newington, T., Vatsiou, A., Fischl, H., Bajracharya, M., Thomson, V. S., Fraser, L. J., Fujita, P. A., Becq, J., Kingsbury, Z., Ross, M. T., Moat, S. J., & Morgan, S. (2023). Whole-Genome Sequencing Can Identify Clinically Relevant Variants from a Single Sub-Punch of a Dried Blood Spot Specimen. International Journal of Neonatal Screening, 9(3), 52. https://doi.org/10.3390/ijns9030052