
Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy

{kind=link}
Abstract
1. Introduction
2. Pathophysiology of Adrenal Insufficiency
3. Epidemiology
4. Clinical Presentation of Adrenal Insufficiency
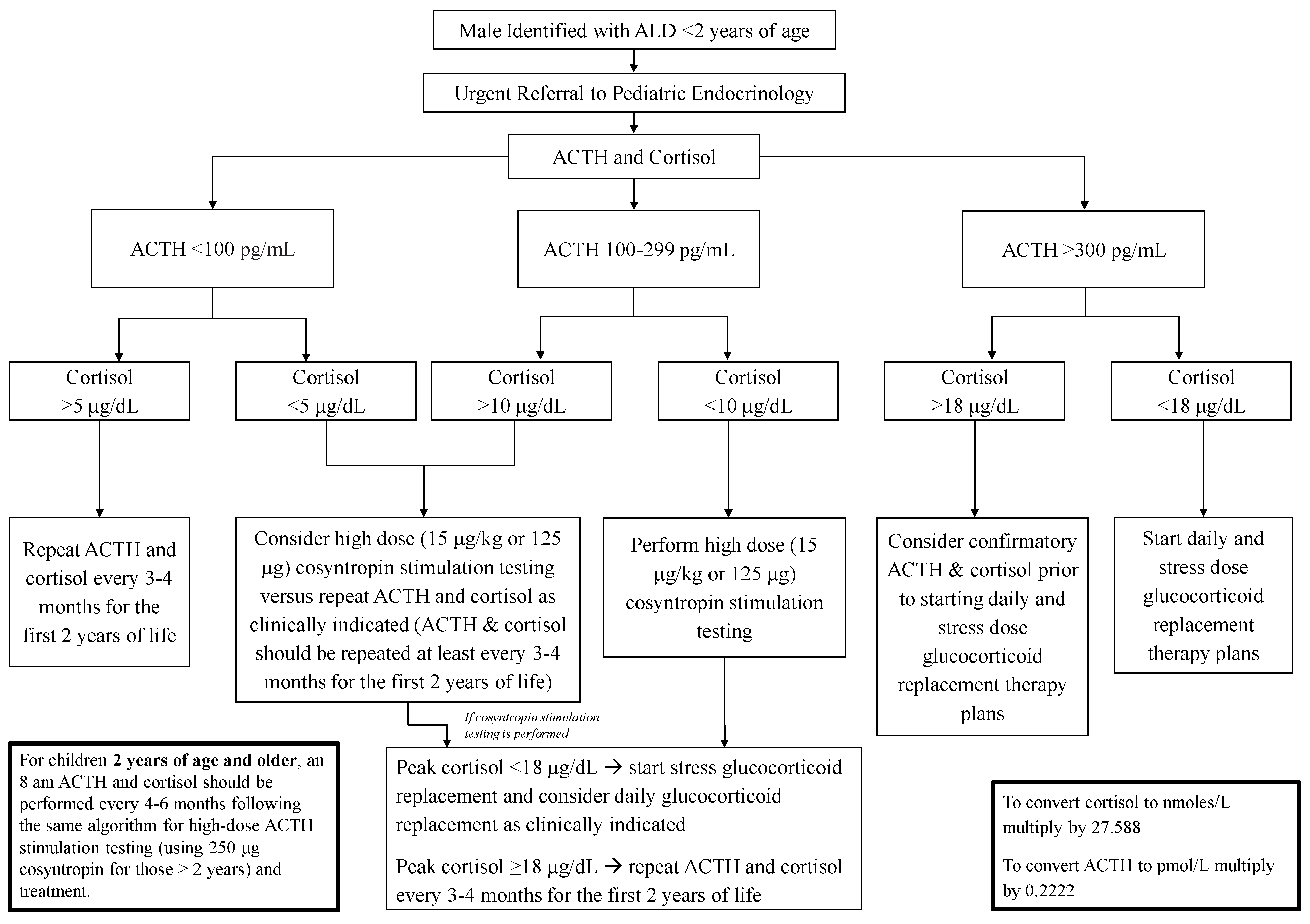
5. Newborn Screening and Adrenal Insufficiency Surveillance
6. Management and Treatment of Adrenal Insufficiency
7. Effects of Investigational Therapies for ALD on Adrenal Function
8. Testicular Dysfunction
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| ALD | Adrenoleukodystrophy |
| AMN | Adrenomyeloneuropathy |
| DHEA | Dehydroepiandrosterone |
| VLCFA | Very long-chain fatty acids |
References
- Raymond, G.V.; Moser, A.B.; Fatemi, A. X-Linked Adrenoleukodystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- ALD Alliance Newborn Screening. Available online: https://www.aldalliance.org/newborn-screening.html (accessed on 30 December 2021).
- Barendsen, R.W.; Dijkstra, I.M.E.; Visser, W.F.; Alders, M.; Bliek, J.; Boelen, A.; Bouva, M.J.; van der Crabben, S.N.; Elsinghorst, E.; van Gorp, A.G.M.; et al. Corrigendum: Adrenoleukodystrophy Newborn Screening in the Netherlands (SCAN Study): The X-Factor. Front. Cell Dev. Biol. 2021, 9, 631655. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Aubourg, P.; Paker, A.; Escolar, M.; Fischer, A.; Blanche, S.; Baruchel, A.; Dalle, J.H.; Michel, G.; Prasad, V.; et al. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; de Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Molzer, B.; Fae, I.; Bernheimer, H. X-linked adrenoleukodystrophy (ALD): A novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem. Biophys. Res. Commun. 1994, 205, 1638–1643. [Google Scholar] [CrossRef]
- Di Rocco, M.; Doria-Lamba, L.; Caruso, U. Monozygotic twins with X-linked adrenoleukodystrophy and different phenotypes. Ann. Neurol. 2001, 50, 424. [Google Scholar] [CrossRef]
- Huffnagel, I.C.; Laheji, F.K.; Aziz-Bose, R.; Tritos, N.A.; Marino, R.; Linthorst, G.E.; Kemp, S.; Engelen, M.; Eichler, F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J. Clin. Endocrinol. Metab. 2019, 104, 118–126. [Google Scholar] [CrossRef]
- Powers, J.M.; Moser, H.W.; Moser, A.B.; Schaumburg, H.H. Fetal adrenoleukodystrophy: The significance of pathologic lesions in adrenal gland and testis. Hum. Pathol. 1982, 13, 1013–1019. [Google Scholar] [CrossRef]
- Powers, J.M.; Schaumburg, H.H.; Johnson, A.B.; Raine, C.S. A correlative study of the adrenal cortex in adreno-leukodystrophy--evidence for a fatal intoxication with very long chain saturated fatty acids. Investig. Cell Pathol. 1980, 3, 353–376. [Google Scholar]
- Whitcomb, R.W.; Linehan, W.M.; Knazek, R.A. Effects of long-chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells In Vitro. J. Clin. Investig. 1988, 81, 185–188. [Google Scholar] [CrossRef]
- Powers, J.M. Adreno-leukodystrophy (adreno-testiculo-leukomyelo-neuropathic-complex). Clin. Neuropathol. 1985, 4, 181–199. [Google Scholar]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.; Schur, R.; de Bie, R.M.; Verhamme, C.; Dijkgraaf, M.G.; Aubourg, P.A.; Wanders, R.J.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain 2014, 137 Pt 3, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Bezman, L.; Moser, A.B.; Raymond, G.V.; Rinaldo, P.; Watkins, P.A.; Smith, K.D.; Kass, N.E.; Moser, H.W. Adrenoleukodystrophy: Incidence, new mutation rate, and results of extended family screening. Ann. Neurol. 2001, 49, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Bonkowsky, J.L.; Wilkes, J.; Bardsley, T.; Urbik, V.M.; Stoddard, G. Association of Diagnosis of Leukodystrophy With Race and Ethnicity Among Pediatric and Adolescent Patients. JAMA Netw. Open 2018, 1, e185031. [Google Scholar] [CrossRef] [PubMed]
- Burtman, E.; Regelmann, M.O. Endocrine Dysfunction in X-Linked Adrenoleukodystrophy. Endocrinol. Metab. Clin. N. Am. 2016, 45, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.; Regelmann, M.O. Adrenoleukodystrophy in the era of newborn screening. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 47–55. [Google Scholar] [CrossRef]
- Regelmann, M.O.; Kamboj, M.K.; Miller, B.S.; Nakamoto, J.M.; Sarafoglou, K.; Shah, S.; Stanley, T.L.; Marino, R. Adrenoleukodystrophy: Guidance for Adrenal Surveillance in Males Identified by Newborn Screen. J. Clin. Endocrinol. Metab. 2018, 103, 4324–4331. [Google Scholar] [CrossRef]
- Matteson, J.; Sciortino, S.; Feuchtbaum, L.; Bishop, T.; Olney, R.S.; Tang, H. Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up. Int. J. Neonatal Screen. 2021, 7, 22. [Google Scholar] [CrossRef]
- Wiens, K.; Berry, S.A.; Choi, H.; Gaviglio, A.; Gupta, A.; Hietala, A.; Kenney-Jung, D.; Lund, T.; Miller, W.; Pierpont, E.I.; et al. A report on state-wide implementation of newborn screening for X-linked Adrenoleukodystrophy. Am. J. Med. Genet. A 2019, 179, 1205–1213. [Google Scholar] [CrossRef]
- Eng, L.; Regelmann, M.O. Early Onset Primary Adrenal Insufficiency in Males with Adrenoleukodystrophy: Case Series and Literature Review. J. Pediatr. 2019, 211, 211–214. [Google Scholar] [CrossRef]
- Dubey, P.; Raymond, G.V.; Moser, A.B.; Kharkar, S.; Bezman, L.; Moser, H.W. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J. Pediatr. 2005, 146, 528–532. [Google Scholar] [CrossRef]
- El-Deiry, S.S.; Naidu, S.; Blevins, L.S.; Ladenson, P.W. Assessment of adrenal function in women heterozygous for adrenoleukodystrophy. J. Clin. Endocrinol. Metab. 1997, 82, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; Eichler, F. Endocrine dysfunction in adrenoleukodystrophy. Handb. Clin. Neurol. 2021, 182, 257–267. [Google Scholar] [PubMed]
- Shulman, D.I.; Palmert, M.R.; Kemp, S.F.; Lawson Wilkins Drug and Therapeutics Committee. Adrenal insufficiency: Still a cause of morbidity and death in childhood. Pediatrics 2007, 119, e484–e494. [Google Scholar] [CrossRef]
- Hoftberger, R.; Kunze, M.; Weinhofer, I.; Aboul-Enein, F.; Voigtlander, T.; Oezen, I.; Amann, G.; Bernheimer, H.; Budka, H.; Berger, J. Distribution and cellular localization of adrenoleukodystrophy protein in human tissues: Implications for X-linked adrenoleukodystrophy. Neurobiol. Dis. 2007, 28, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Oelkers, W.; Boelke, T.; Bahr, V. Dose-response relationships between plasma adrenocorticotropin (ACTH), cortisol, aldosterone, and 18-hydroxycorticosterone after injection of ACTH-(1-39) or human corticotropin-releasing hormone in man. J. Clin. Endocrinol. Metab. 1988, 66, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.H.; Bradley, S.E.; Adams, D.J.; D’Aco, K.; Erbe, R.W.; Fong, C.; Iglesias, A.; Kronn, D.; Levy, P.; Morrissey, M.; et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: Diagnostic protocol, surveillance protocol and treatment guidelines. Mol. Genet. Metab. 2015, 114, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Klouwer, F.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.; Engelen, M.; Poll-The, B.T. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare Dis. 2015, 10, 151. [Google Scholar] [CrossRef]
- Wang, R.Y.; Monuki, E.S.; Powers, J.; Schwartz, P.H.; Watkins, P.A.; Shi, Y.; Moser, A.; Shrier, D.A.; Waterham, H.R.; Nugent, D.J.; et al. Effects of hematopoietic stem cell transplantation on acyl-CoA oxidase deficiency: A sibling comparison study. J. Inherit. Metab. Dis. 2014, 37, 791–799. [Google Scholar] [CrossRef]
- Chapel-Crespo, C.C.; Villalba, R.; Wang, R.; Boyer, M.; Chang, R.; Waterham, H.R.; Abdenur, J.E. Primary adrenal insufficiency in two siblings with D-bifunctional protein deficiency. Mol. Genet. Metab. Rep. 2020, 24, 100608. [Google Scholar] [CrossRef]
- Stahl, F.; Amendt, P.; Dorner, G. Total and free cortisol plasma levels in pre- and postnatal life. Endokrinologie 1979, 74, 243–246. [Google Scholar]
- Javorsky, B.R.; Raff, H.; Carroll, T.B.; Algeciras-Schimnich, A.; Singh, R.J.; Colon-Franco, J.M.; Findling, J.W. New Cutoffs for the Biochemical Diagnosis of Adrenal Insufficiency after ACTH Stimulation using Specific Cortisol Assays. J. Endocr. Soc. 2021, 5, bvab022. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Allolio, B.; Arlt, W.; Barthel, A.; Don-Wauchope, A.; Hammer, G.D.; Husebye, E.S.; Merke, D.P.; Murad, M.H.; Stratakis, C.A.; et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 364–389. [Google Scholar] [CrossRef] [PubMed]
- Ngaosuwan, K.; Johnston, D.G.; Godsland, I.F.; Cox, J.; Majeed, A.; Quint, J.K.; Oliver, N.; Robinson, S. Mortality Risk in Patients With Adrenal Insufficiency Using Prednisolone or Hydrocortisone: A Retrospective Cohort Study. J. Clin. Endocrinol. Metab. 2021, 106, 2242–2251. [Google Scholar] [CrossRef]
- Miller, B.S.; Spencer, S.P.; Geffner, M.E.; Gourgari, E.; Lahoti, A.; Kamboj, M.K.; Stanley, T.L.; Uli, N.K.; Wicklow, B.A.; Sarafoglou, K. Emergency management of adrenal insufficiency in children: Advocating for treatment options in outpatient and field settings. J. Investig. Med. 2020, 68, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kloesel, B.; Dua, N.; Eskuri, R.; Hall, J.; Cohen, M.; Richtsfeld, M.; Belani, K. Anesthetic management of pediatric patients diagnosed with X-linked adrenoleukodystrophy: A single-center experience. Paediatr. Anaesth. 2020, 30, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.; Barker, P.; Daniel, S.; Fletcher, S.; Wass, J.A.H.; Tomlinson, J.W.; Misra, U.; Dattani, M.; Arlt, W.; Vercueil, A. Guidelines for the management of glucocorticoids during the peri-operative period for patients with adrenal insufficiency: Guidelines from the Association of Anaesthetists, the Royal College of Physicians and the Society for Endocrinology UK. Anaesthesia 2020, 75, 654–663. [Google Scholar] [CrossRef]
- Mallack, E.J.; Turk, B.R.; Yan, H.; Price, C.; Demetres, M.; Moser, A.B.; Becker, C.; Hollandsworth, K.; Adang, L.; Vanderver, A.; et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J. Inherit. Metab. Dis. 2021, 44, 728–739. [Google Scholar] [CrossRef]
- Petryk, A.; Polgreen, L.E.; Chahla, S.; Miller, W.; Orchard, P.J. No evidence for the reversal of adrenal failure after hematopoietic cell transplantation in X-linked adrenoleukodystrophy. Bone Marrow Transplant. 2012, 47, 1377–1378. [Google Scholar] [CrossRef]
- FDA Halts Trial of Gene Therapy for Rare Neurological Disease Due to Cancer Risk. Available online: https://www.healio.com/news/hematology-oncology/20210812/fda-halts-trial-of-gene-therapy-for-rare-neurological-disease-due-to-cancer-risk (accessed on 30 December 2021).
- Uziel, G.; Bertini, E.; Bardelli, P.; Rimoldi, M.; Gambetti, M. Experience on therapy of adrenoleukodystrophy and adrenomyeloneuropathy. Dev. Neurosci. 1991, 13, 274–279. [Google Scholar] [CrossRef]
- Aubourg, P.; Adamsbaum, C.; Lavallard-Rousseau, M.C.; Rocchiccioli, F.; Cartier, N.; Jambaque, I.; Jakobezak, C.; Lemaitre, A.; Boureau, F.; Wolf, C.; et al. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy. N. Engl. J. Med. 1993, 329, 745–752. [Google Scholar] [CrossRef]
- Rizzo, W.B.; Leshner, R.T.; Odone, A.; Dammann, A.L.; Craft, D.A.; Jensen, M.E.; Jennings, S.S.; Davis, S.; Jaitly, R.; Sgro, J.A. Dietary erucic acid therapy for X-linked adrenoleukodystrophy. Neurology 1989, 39, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, B.M.; Assies, J.; Haverkort, E.B.; Koelman, J.H.; Verbeeten, B., Jr.; Wanders, R.J.; Barth, P.G. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with “Lorenzo’s oil”. J. Neurol. Neurosurg. Psychiatry 1999, 67, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Moser, H.W.; Raymond, G.V.; Koehler, W.; Sokolowski, P.; Hanefeld, F.; Korenke, G.C.; Green, A.; Loes, D.J.; Hunneman, D.H.; Jones, R.O.; et al. Evaluation of the preventive effect of glyceryl trioleate-trierucate ("Lorenzo’s oil") therapy in X-linked adrenoleukodystrophy: Results of two concurrent trials. Adv. Exp. Med. Biol. 2003, 544, 369–387. [Google Scholar]
- Cappa, M.; Bizzarri, C.; Giannone, G.; Aiello, C.; di Biase, A. Is subclinical adrenal failure in adrenoleukodystrophy/adrenomyeloneuropathy reversible? J. Endocrinol. Investig. 2011, 34, 753–756. [Google Scholar]
- Assies, J.; Gooren, L.J.; van Geel, B.; Barth, P.G. Signs of testicular insufficiency in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy: A retrospective study. Int. J. Androl. 1997, 20, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Brennemann, W.; Kohler, W.; Zierz, S.; Klingmuller, D. Testicular dysfunction in adrenomyeloneuropathy. Eur. J. Endocrinol. 1997, 137, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Stradomska, T.J.; Kubalska, J.; Janas, R.; Tylki-Szymanska, A. Reproductive function in men affected by X-linked adrenoleukodystrophy/adrenomyeloneuropathy. Eur. J. Endocrinol. 2012, 166, 291–294. [Google Scholar] [CrossRef]
- Powers, J.M.; Schaumburg, H.H. The testis in adreno-leukodystrophy. Am. J. Pathol. 1981, 102, 90–98. [Google Scholar]
- Karapanou, O.; Vlassopoulou, B.; Tzanela, M.; Papadopoulos, D.; Angelidakis, P.; Michelakakis, H.; Ioannidis, G.; Mihalatos, M.; Kamakari, S.; Tsagarakis, S. X-linked adrenoleukodystrophy: Are signs of hypogonadism always due to testicular failure? Hormones 2014, 13, 146–152. [Google Scholar] [CrossRef]
- Assies, J.; Haverkort, E.B.; Lieverse, R.; Vreken, P. Effect of dehydroepiandrosterone supplementation on fatty acid and hormone levels in patients with X-linked adrenoleucodystrophy. Clin. Endocrinol. 2003, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kachwala, I.; Regelmann, M.O. Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy. Int. J. Neonatal Screen. 2022, 8, 18. https://doi.org/10.3390/ijns8010018
Kachwala I, Regelmann MO. Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy. International Journal of Neonatal Screening. 2022; 8(1):18. https://doi.org/10.3390/ijns8010018
Chicago/Turabian StyleKachwala, Isha, and Molly O. Regelmann. 2022. "Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy" International Journal of Neonatal Screening 8, no. 1: 18. https://doi.org/10.3390/ijns8010018
APA StyleKachwala, I., & Regelmann, M. O. (2022). Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy. International Journal of Neonatal Screening, 8(1), 18. https://doi.org/10.3390/ijns8010018