
Massachusetts’ Findings from Statewide Newborn Screening for Spinal Muscular Atrophy

 ,
,
Abstract
1. Introduction
2. Materials and Methods
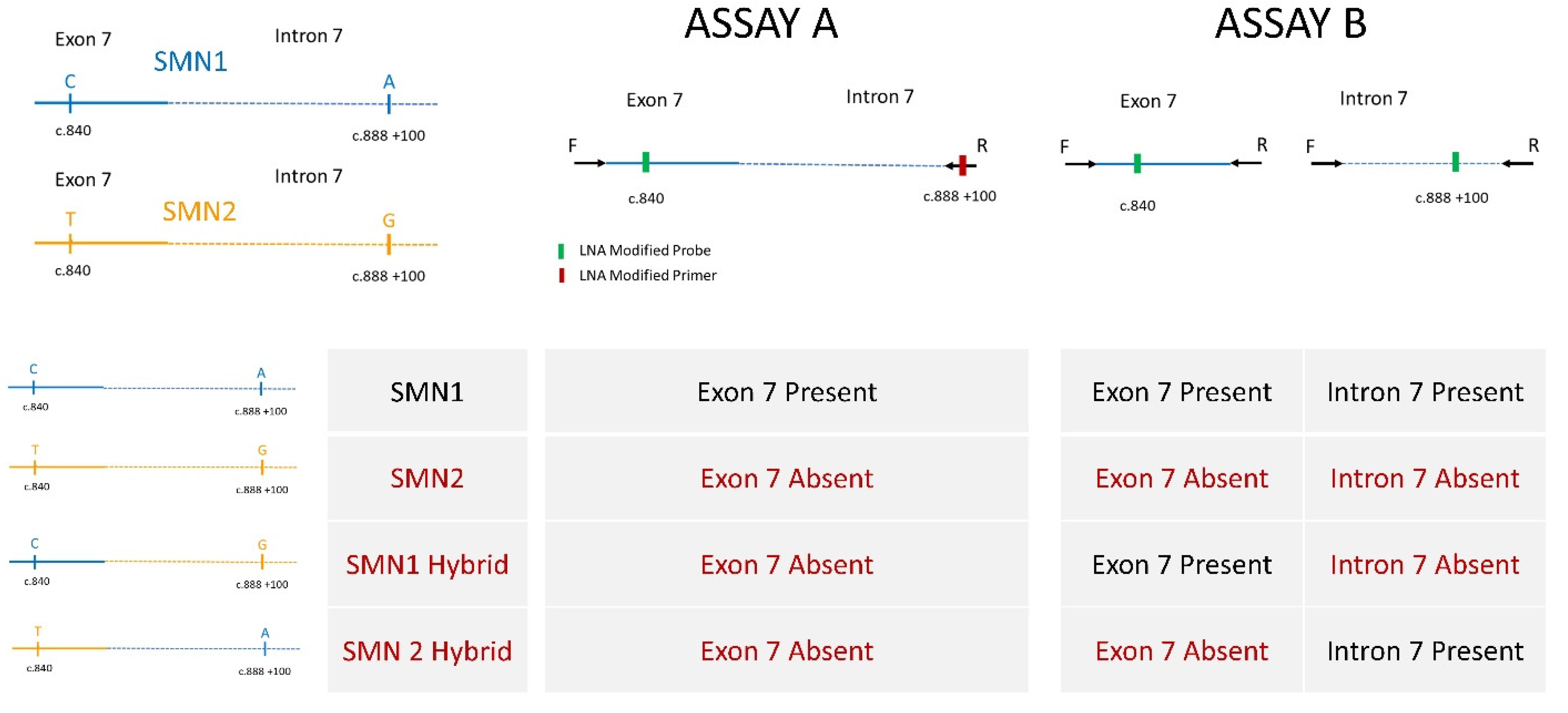
2.1. Assay Development
2.2. DNA Preparation for High-Throughput Assay
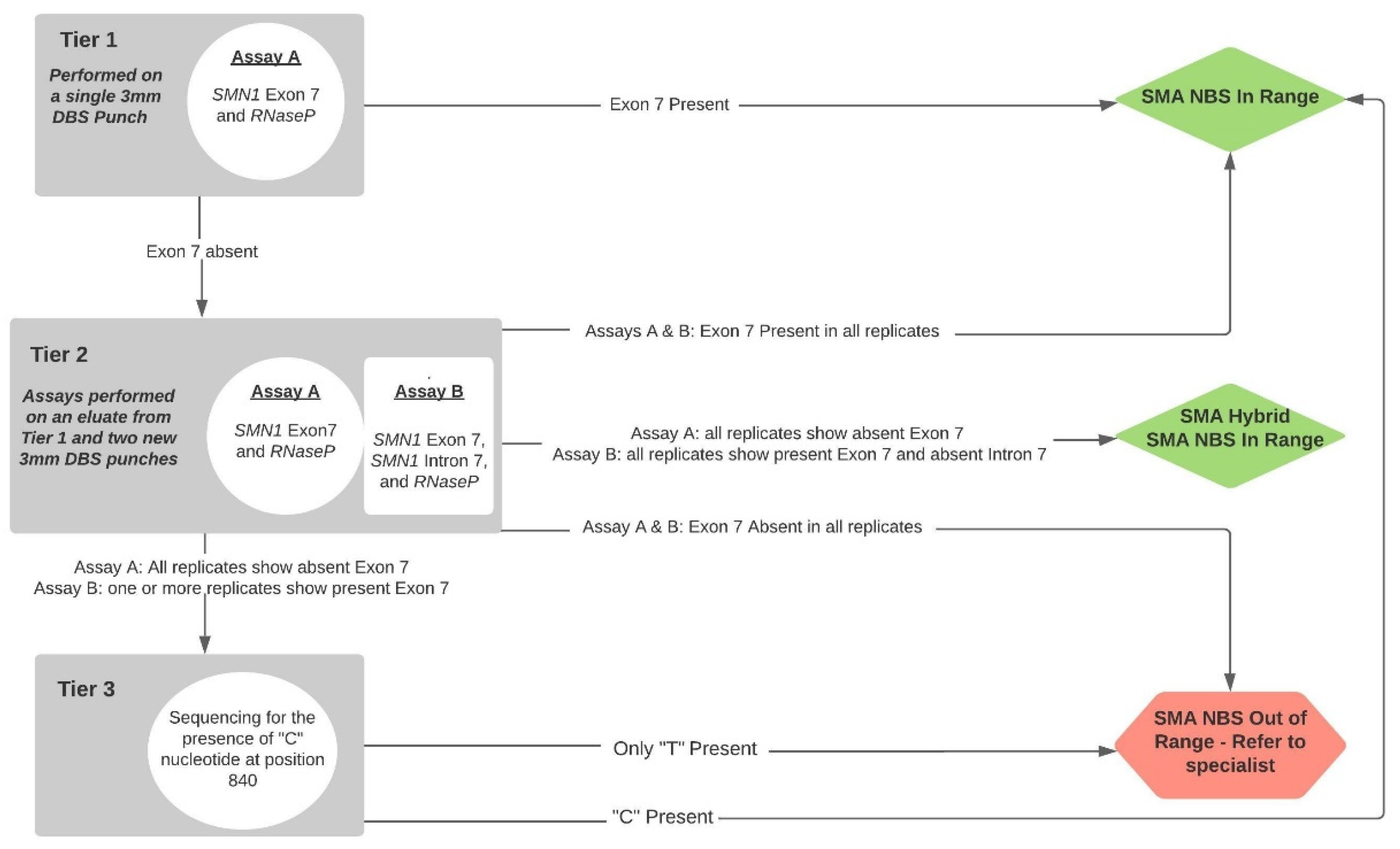
2.3. Statewide Screening Implementation
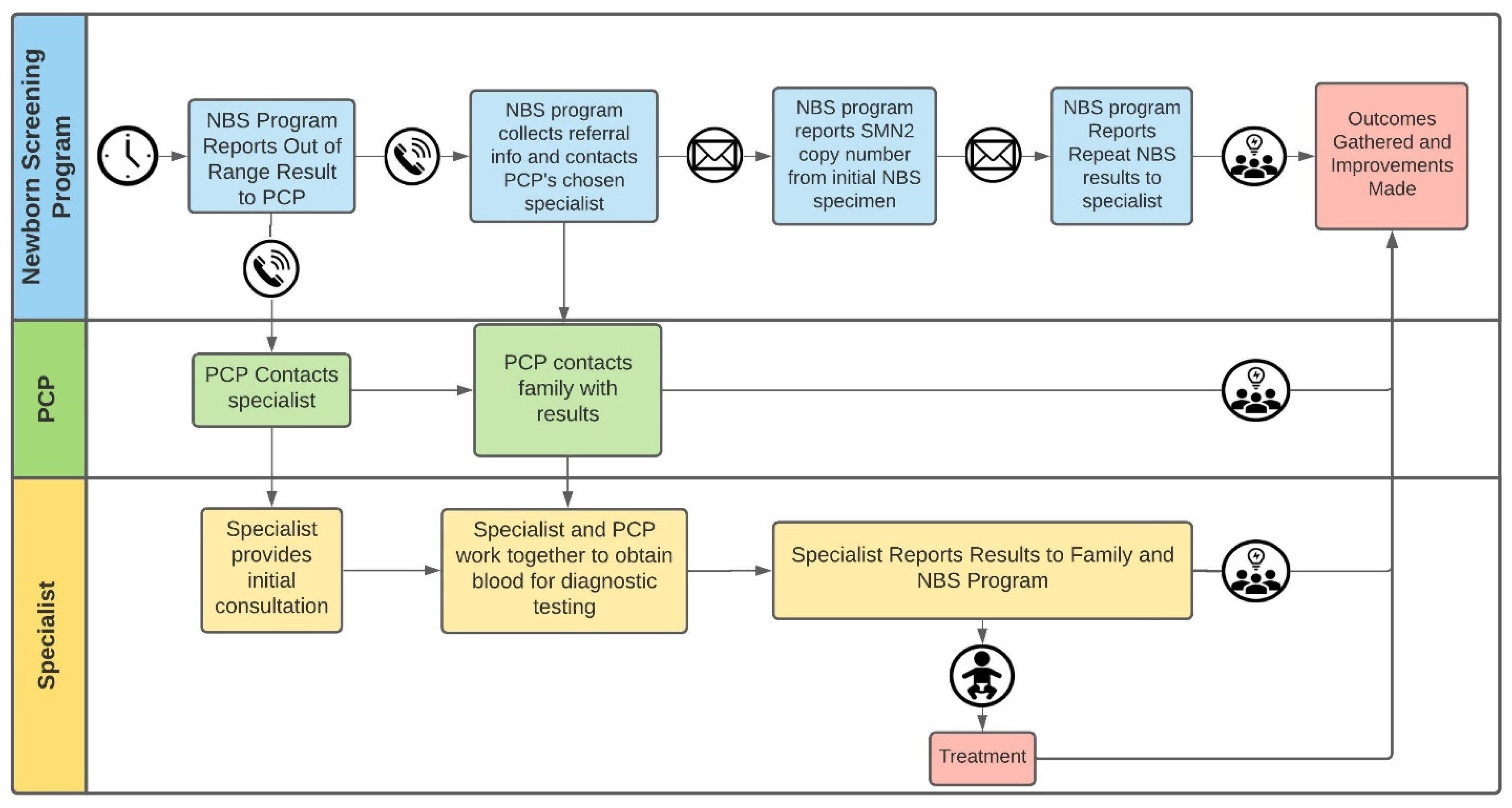
2.4. Referrals and Outcomes Analyses
3. Results
3.1. Screening
3.2. Outcomes of Infants Who Were Not Referred, Whose Specimens Initially Showed Absence of SMN1 Due to Presence of SMN1 Hybrid Gene
3.3. Screening Data and Short-Term Clinical Outcomes of the Nine SMA-Affected Infants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J. Pediatr. 2017, 190, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Advisory Committee on Heritable Disorders in Newborns and Children. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/reports-recommendations/final-sign-azar-response-sma.pdf (accessed on 19 March 2021).
- NewSTEPs: A Program of the Association of Public Health Laboratories. Available online: https://www.newsteps.org/resources/newborn-screening-status-all-disorders (accessed on 19 March 2021).
- Taylor, J.L.; Lee, F.K.; Yazdanpanah, G.K.; Staropoli, J.F.; Liu, M.; Carulli, J.P.; Sun, C.; Dobrowolski, S.F.; Hannon, W.H.; Vogt, R.F. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin. Chem. 2015, 61, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K. Newborn screening for spinal muscular atrophy. In Proceedings of the Newborn Screening and Genetic Testing Symposium, New Orleans, LA, USA, 10–13 September 2017; Available online: https://www.aphl.org/conferences/proceedings/Documents/Mercer.pdf (accessed on 27 April 2021).
- Lee, F.K. Newborn screening for spinal muscular atrophy (SMA) in the US. In Proceedings of the Spinal Muscular Atrophy: Overview of Available Screening Methods, Webinar. 28 June 2018; Available online: https://www.newsteps.org/sites/default/files/resources/download/aphl_sma_webinar_slides_webinarslides_june2018_kh.pdf (accessed on 27 April 2021).
- Gerstel-Thompson, J.L.; Wilkey, J.F.; Baptiste, J.C.; Navas, J.S.; Pai, S.Y.; Pass, K.A.; Eaton, R.B.; Comeau, A.M. High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin. Chem. 2010, 56, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Zytkovicz, T.H.; Fitzgerald, E.F.; Marsden, D.; Larson, C.A.; Shih, V.E.; Johnson, D.M.; Strauss, A.W.; Comeau, A.M.; Eaton, R.B.; Grady, G.F. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: A two-year summary from the New England Newborn Screening Program. Clin. Chem. 2001, 47, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Parad, R.B.; Dorkin, H.L.; Dovey, M.; Gerstle, R.; Haver, K.; Lapey, A.; O’Sullivan, B.P.; Waltz, D.A.; Zwerdling, R.G.; et al. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: A cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics 2004, 113, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Hale, J.E.; Pai, S.Y.; Bonilla, F.A.; Notarangelo, L.D.; Pasternack, M.S.; Meissner, H.C.; Cooper, E.R.; DeMaria, A.; Sahai, I.; et al. Guidelines for implementation of population-based newborn screening for severe combined immunodeficiency. J. Inherit. Metab. Dis. 2010, 33, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Connolly, A.M.; Darras, B.T.; Day, J.W.; Finkel, R.; Howell, R.R.; Klinger, K.W.; Kuntz, N.; Prior, T.; et al. Revised Recommendations for the Treatment of Infants Diagnosed with Spinal Muscular Atrophy Via Newborn Screening Who Have 4 Copies of SMN2. J. Neuromuscul. Dis. 2020, 7, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—A literature review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Carrier Screening for Genetic Conditions. Committee Opinion No. 691. American College of Obstetricians and Gynecologists. Obstet. Gynecol. 2017, 129, e41–e55. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Infants Whose Specimens | SMA NBS Interpretation | Number of Infants |
|---|---|---|
| Showed Exon 7 to be present in Tier 1 | In Range | 179,153 |
| Showed Exon 7 to be absent in Tier 1 | ||
| Showed absent Exon 7 in Tier 2 | Out of Range | 10 1 |
| Showed evidence of an SMN1 hybrid in Tier 2 | In Range | 10 |
| Showed evidence of present SMN1 in Tier 2 | In Range | 294 |
| TOTAL SCREENED | 179,467 |
| Age (Months) at Follow up Call | Age (Months) at Last Visit to PCP | Reported Status |
|---|---|---|
| 17.8 | 16.0 | very healthy, runs around the room, no neuromuscular clinical concerns |
| 14.6 | 12.0 | no neuromuscular clinical concerns; does have unrelated genetic diagnosis |
| 14.0 | 12.0 | well and no neuromuscular clinical concerns |
| 9.1 | 6.4 | no neuromuscular clinical concerns; umbilical hernia |
| 7.7 | Not reported | well and no neuromuscular clinical concerns |
| 6.8 | 3.7 | well child visit; normal strength and tone; growth 75th percentile |
| Case | SMN2 Copy Number (from Diagnostic Lab) | Age at Specimen Collection/Receipt by NBS/When Screening Result Communicated (Days of Life) | Prenatal Testing | Clinical Status Reported by PCP at Time of NBS Report | Age at First Visit to Specialist (Days of Life) | Clinical Status Reported by Specialist at First Evaluation | Treatment Type | Age at First Treatment (Days of Life) | Current Age (Months) | Current Clinical Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 2/2/3 | Both parents known carriers | Well | 26 | No symptoms | Nusinersen | 38 | 24 | Normal developmental progression for age |
| 2 | 4 | 2/4/5 | Well; gaining weight but not quite back up to birthweight | 5 | Tongue fasciculations and mild generalized hypotonia | Onasemnogene abeparvovec | 171 | 22 | Continues to develop normally without symptoms or signs of SMA | |
| 3 | 2 | 2/4/4 | Both parents known carriers | Poor tone | 6 | Hypotonia, decreased movements, head lag and slip through on exam; Respiratory insufficiency, bell shaped chest, paradoxical breathing | Onasemnogene abeparvovec | 18 | 19.5 | Moderate motor delays; sits unassisted, rolls over, moderate head control, cannot lift head from a prone position; able to transfer objects hand to hand. Cannot bear full weight on legs, no crawling or walkingAble to chew and swallow, no secretion problems |
| 4 | 2 | 1/3/6 | Well | 9 | Day of life (DOL) 7 normal echo and microcephaly per medical records and + axillary slip; DOL 9 tongue fasciculations and impaired swallowing, generalized weakness and hypotonia | Nusinersen/Onasemnogene abeparvovec/Risdiplam | 16/29/309 | 19 | At 15 months, crawling, sitting independently, pulls to stand; walks with a walker. Although required g-tube initially, now 100% oral feeder | |
| 5 | 2 | 1/2/3 | Premature but well; no SMA concerns | 7 | Paradoxical breathing and tongue fasciculations | Nusinersen/Onasemnogene abeparvovec | 11/98 | 13 | At 1 years old, continues to acquire age-appropriate motor milestones: rolling, sitting unsupported, crawling | |
| 6 | 2 | 2/3/6 | Extremely dislocatable left hip, low muscle tone | 8 | No symptoms | Onasemnogene abeparvovec | 29 | 12 | Mild motor delays (probably in part due to bracing for hip dislocation) | |
| 7 | 4 | 1/2/3 | Prenatal diagnosis | Status reported as already under the care of a specialist | 0 | No symptoms | Nusinersen | 8 | 10 | Remains asymptomatic at 9 months of age |
| 8 | 2 | 2/2/3 | One parent known carrier | Well | 13 | Tongue fasciculations and reduced deep tendon reflexes | Onasemnogene abeparvovec | 13 | 5.5 | At 4 months, continues to acquire improved tone and head control in prone and supported sitting positions and recently began eating purees |
| 9 | 2 | 2/5/6 | Both parents known carriers | Well | 7 | Limited movements of bilateral lower extremities | Onasemnogene abeparvovec | 19 | 5 | Weakness of bilateral lower extremities left > right |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hale, J.E.; Darras, B.T.; Swoboda, K.J.; Estrella, E.; Chen, J.Y.H.; Abbott, M.-A.; Hay, B.N.; Kumar, B.; Counihan, A.M.; Gerstel-Thompson, J.; et al. Massachusetts’ Findings from Statewide Newborn Screening for Spinal Muscular Atrophy. Int. J. Neonatal Screen. 2021, 7, 26. https://doi.org/10.3390/ijns7020026
Hale JE, Darras BT, Swoboda KJ, Estrella E, Chen JYH, Abbott M-A, Hay BN, Kumar B, Counihan AM, Gerstel-Thompson J, et al. Massachusetts’ Findings from Statewide Newborn Screening for Spinal Muscular Atrophy. International Journal of Neonatal Screening. 2021; 7(2):26. https://doi.org/10.3390/ijns7020026
Chicago/Turabian StyleHale, Jaime E., Basil T. Darras, Kathryn J. Swoboda, Elicia Estrella, Jin Yun Helen Chen, Mary-Alice Abbott, Beverly N. Hay, Binod Kumar, Anne M. Counihan, Jacalyn Gerstel-Thompson, and et al. 2021. "Massachusetts’ Findings from Statewide Newborn Screening for Spinal Muscular Atrophy" International Journal of Neonatal Screening 7, no. 2: 26. https://doi.org/10.3390/ijns7020026
APA StyleHale, J. E., Darras, B. T., Swoboda, K. J., Estrella, E., Chen, J. Y. H., Abbott, M.-A., Hay, B. N., Kumar, B., Counihan, A. M., Gerstel-Thompson, J., Sahai, I., Eaton, R. B., & Comeau, A. M. (2021). Massachusetts’ Findings from Statewide Newborn Screening for Spinal Muscular Atrophy. International Journal of Neonatal Screening, 7(2), 26. https://doi.org/10.3390/ijns7020026