Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy

 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. LSD Screening Assay
2.3. Second Tier Tests
2.4. Confirmatory Testing
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P.C.J.I. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn screening for lysosomal storage diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Diagnosis of α-l-iduronidase deficiency in dried blood spots on filter paper: the possibility of newborn diagnosis. Clin. Chem. 2001, 47, 780–781. [Google Scholar] [PubMed]
- Gelb, M.H.; Turecek, F.; Scott, C.R.; Chamoles, N.A. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J. Inherit. Metab. Dis. 2006, 29, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Sista, R.S.; Eckhardt, A.E.; Wang, T.; Graham, C.; Rouse, J.L.; Norton, S.M.; Srinivasan, V.; Pollack, M.G.; Tolun, A.A.; Bali, D.; et al. Digital Microfluidic Platform for Multiplexing Enzyme Assays: Implications for Lysosomal Storage Disease Screening in Newborns. Clin. Chem. 2011, 57, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tükel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Chiang, S.-C.; Zhang, X.K.; Keutzer, J.; Lee, N.-C.; Huang, A.-C.; Chen, C.-A.; Wu, M.-H.; Huang, P.-H.; Tsai, F.-J.; et al. Early detection of Pompe disease by newborn screening is feasible: Results from the Taiwan screening program. Pediatrics 2008, 122, e39–e45. [Google Scholar] [CrossRef]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for fabry disease in Taiwan reveals a high incidence of the later-onset mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2010, 30, 1397–1405. [Google Scholar] [CrossRef]
- Duffner, P.K.; Caggana, M.; Orsini, J.J.; Wenger, D.A.; Patterson, M.C.; Crosley, C.J.; Kurtzberg, J.; Arnold, G.L.; Escolar, M.L.; Adams, D.J.; et al. Newborn screening for Krabbe disease: The New York State model. Pediatr. Neurol. 2009, 40, 245–252. [Google Scholar] [CrossRef]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn screening for lysosomal storage disorders in Illinois: The initial 15-month experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the first 65,000 infants. Genet. Med. 2018, 21, 631. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html (accessed on 27 March 2019).
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019. [Google Scholar] [CrossRef]
- Johnson, B.; Mascher, H.; Mascher, D.; Legnini, E.; Hung, C.Y.; Dajnoki, A.; Chien, Y.H.; Maródi, L.; Hwu, W.L.; Bodamer, O.A. Analysis of lyso-globotriaosylsphingosine in dried blood spots. Ann. Lab. Med. 2013, 33, 274–278. [Google Scholar] [CrossRef]
- De Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef]
- Regione Veneto, Health Department. Available online: https://bur.regione.veneto.it/BurvServices/pubblica/DettaglioDgr.aspx?id=254110 (accessed on 17 May 2019).
- Regione Friuli Venezia Giulia, Health Department. Available online: http://mtom.regione.fvg.it/storage//2016_74/Testo integrale della Delibera n 74-2016.pdf (accessed on 17 May 2019).
- Zhang, H.; Young, S.P.; Auray-Blais, C.; Orchard, P.J.; Tolar, J.; Millington, D.S. Analysis of glycosaminoglycans in cerebrospinal fluid from patients with mucopolysaccharidoses by Isotope-dilution ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2011, 57, 1005–1012. [Google Scholar] [CrossRef]
- Rozaklis, T.; Ramsay, S.L.; Whitfield, P.D.; Ranieri, E.; Hopwood, J.J.; Meikle, P.J. Determination of oligosaccharides in Pompe disease by electrospray ionization tandem mass spectrometry. Clin. Chem. 2002, 48, 131–139. [Google Scholar]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of sphingolipidoses: A new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Bherer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Turaca, L.T.; de Faria, D.O.; Kyosen, S.O.; Teixeira, V.D.; Motta, F.L.; Pessoa, J.G.; e Silva, M.R.; de Almeida, S.S.; D’Almeida, V.; Rojas, M.V.; et al. Novel GAA mutations in patients with Pompe disease. Gene 2015, 561, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.E.; Amartino, H.M.; Niizawa, G.; Blanco, M.; Pomponio, R.J.; Chamoles, N.A. Pompe disease (glycogen storage disease type II) in Argentineans: Clinical manifestations and identification of 9 novel mutations. Neuromuscul Disord. 2007, 17, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Zanoutene-Cheriet, L.; Adjtoutah, Z.; Abily-Donval, L.; Brasse-Lagnel, C.; Laquerrière, A.; Marret, S.; Benabdellah, A.C.; Bekri, S.; Tikkanen, R. Clinical and molecular characterization of patients with mucopolysaccharidosis type I in an algerian series. Int. J. Mol. Sci. 2016, 17, 743. [Google Scholar] [CrossRef] [PubMed]
- Al-Sannaa, N.A.; Bay, L.; Barbouth, D.S.; Benhayoun, Y.; Goizet, C.; Guelbert, N.; Jones, S.A.; Kyosen, S.O.; Martins, A.M.; Phornphutkul, C.; et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet J. Rare Dis. 2015, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Filocamo, M.; Mazzotti, R.; Stroppiano, M.; Seri, M.; Giona, F.; Parenti, G.; Regis, S.; Corsolini, F.; Zoboli, S.; Gatti, R. Analysis of the glucocerebrosidase gene and mutationprofile in 144 Italiangaucherpatients. Hum. Mutat. 2002, 20, 234–235. [Google Scholar] [CrossRef]
- International Fabry Disease Genotype-Phenotype Database (dbFGP). Available online: http://www.dbfgp.org/dbFgp/fabry/FabryGP.html (accessed on 27 March 2019).
- Dionisi-Vici, C.; Rizzo, C.; Burlina, A.B.; Caruso, U.; Sabetta, G.; Uziel, G.; Abeni, D. Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 2002, 140, 321–329. [Google Scholar] [CrossRef]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 126. [Google Scholar] [CrossRef]
- Gelb, M.H. Newborn screening for lysosomal storage diseases: Methodologies, screen positive rates, normalization of datasets, second-tier tests, and post-analysis tools. Int. J. Neonatal Screen. 2018, 4, 23. [Google Scholar] [CrossRef]
- Pettazzoni, M.; Froissart, R.; Pagan, C.; Vanier, M.T.; Ruet, S.; Latour, P.; Guffon, N.; Fouilhoux, A.; Germain, D.P.; Levade, T.; et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS ONE 2017, 12, e0181700. [Google Scholar] [CrossRef]
- Smid, B.E.; van der Tol, L.; Biegstraaten, M.; Linthorst, G.E.; Hollak, C.E.M.; Poorthuis, B.J.H.M. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J. Med. Genet. 2015, 52, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Kasper, D.; Pagliardini, V.; Biamino, E.; Giachero, S.; Porta, F. Metabolic progression to clinical phenotype in classic Fabry disease. Ital. J. Pediatr. 2017, 43, 1. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Bodamer, O.A.; Chiang, S.C.; Mascher, H.; Hung, C.; Hwu, W.L. Lyso-globotriaosylsphingosine (lyso-Gb3) levels in neonates and adults with the Fabry disease later-onset GLA IVS4+919G>A mutation. J. Inherit. Metab. Dis. 2013, 36, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Suzuki, Y.; Orii, K.; Giugliani, R.; Church, H.J.; Mason, R.W.; Dũng, V.C.; Ngoc, C.T.B.; Yamaguchi, S.; Kobayashi, H.; et al. Glycosaminoglycan levels in dried blood spots of patients with mucopolysaccharidoses and mucolipidoses. Mol. Genet. Metab. 2017, 120, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Macklin, S.; Laney, D.; Lisi, E.; Atherton, A.; Smith, E. The psychosocial impact of carrying a debated variant in the GLA gene. J. Genet. Couns. 2018, 27, 217–224. [Google Scholar] [CrossRef] [PubMed]


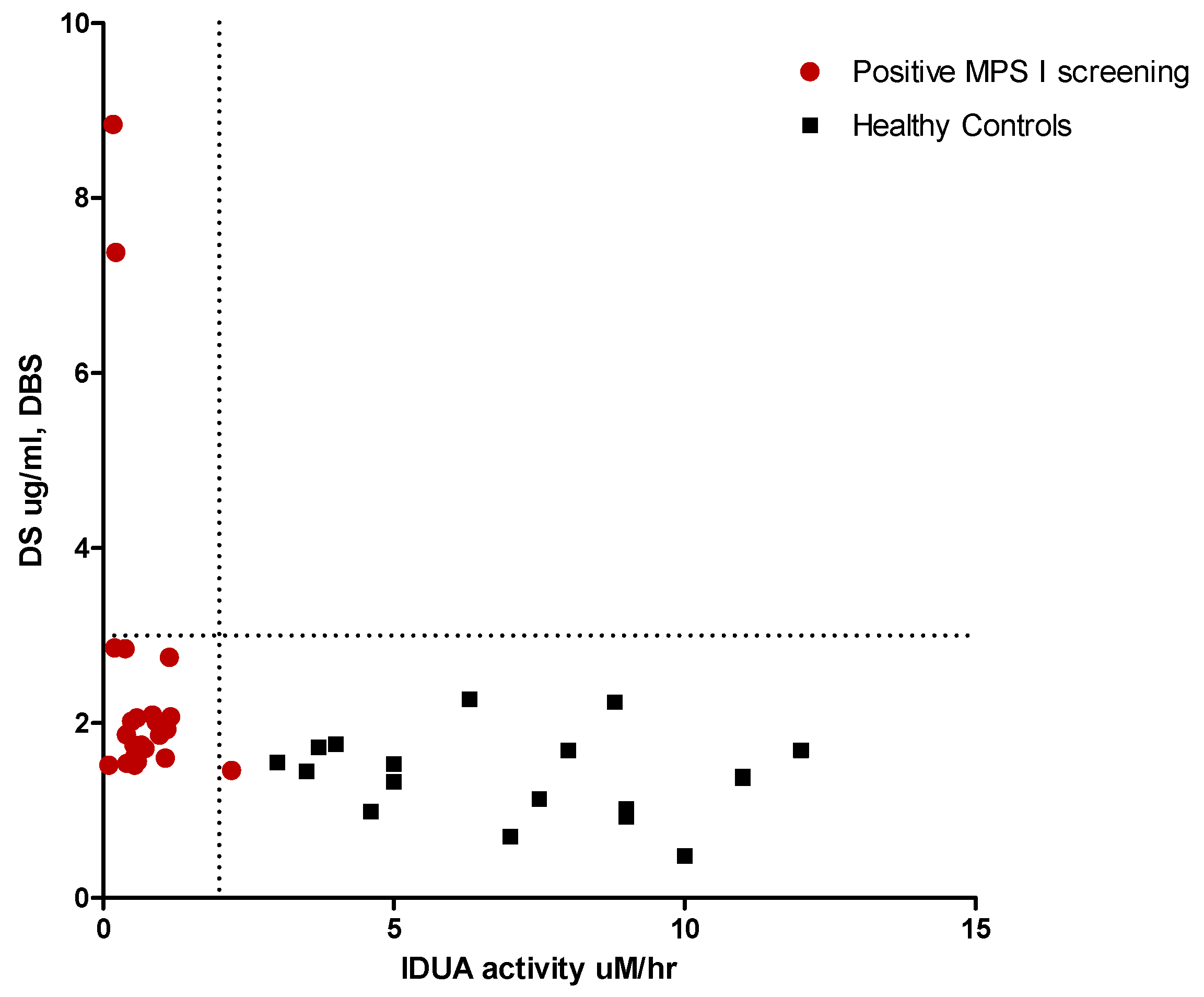{kind=link}
| LSD | Total Screened | Positive NBS | Recall Rate | Patient Undergo to Confirmatory Testing | Patients with Confirmed Disorder | Pseudo Deficit | VUS | Carrier Status |
|---|---|---|---|---|---|---|---|---|
| Gaucher | 112,446 | 28 | 0.03 | 7 | 7 | 0 | 0 | 0 |
| Pompe | 112,446 | 28 | 0.03 | 18 | 8 | 7 | 2 | 1 |
| Fabry | 112,446 | 23 | 0.02 | 11 | 8 | 1 | 2 | 0 |
| MPS I | 112,446 | 52 | 0.05 | 26 | 2 | 20 | 2 | 2 |
| Multiple LSD | 112,446 | 7 | 0.01 | 0 | ||||
| Total | 112,446 | 138 | 0.12 | 62 | 25 | 28 | 6 | 3 |
| Pompe Disease | NBS Enzyme Activity | % Enzyme Activity | Sex | Ethnic Origin | Biochemical Test * | Glc4 ** (mmol/mol creat) | Genotype | Predicted Phenotype |
|---|---|---|---|---|---|---|---|---|
| GAA-01 | 0.2 | 1.4 | M | West Africa | CPK 1063 ALT 15 AST 112 | 71.2 | p.R854X/NF | IOPD |
| GAA-02 | 0.45 | 3.1 | F | European | CPK 990 ALT 69 AST 128 | 94.4 | p.D645N/p.W746X | IOPD |
| GAA-03 | 0.61 | 4.2 | M | North Africa | CPK 448 ALT 48 AST 98 | NP | IVS1-13T>G/p.P79RfsX12 | LOPD |
| GAA-04 | 1.03 | 7.1 | M | European | Normal | 0.5 | IVS1-13T>G/IVS1-13T>G | LOPD |
| GAA-05 | 1.11 | 7.6 | M | European | Normal | NP | IVS1-13T>G/IVS1-13T>G | LOPD |
| GAA-06 | 1.17 | 8.0 | M | European | Normal | 0.2 | IVS1-13T>G/IVS1-13T>G | LOPD |
| GAA-07 | 1.41 | 9.7 | F | North Africa | CPK 682 ALT 68 AST 130 | 20.1 | IVS1-13T>G/p.P79RfsX12 | LOPD |
| GAA-08 | 1.43 | 9.8 | M | European | Normal | NP | p.V222M/p.V222M | Pseudo |
| GAA-09 | 1.84 | 12.6 | M | European | NP | NP | p.R385H_p.E689K_p.A704T_p.W746C | Pseudo |
| GAA-10 | 1.89 | 13.0 | M | Asia | Normal | NP | p.D645E/wt | Carrier PD |
| GAA-11 | 1.92 | 13.2 | M | Asia | Normal | NP | p.G576S/p.E689K | Pseudo |
| GAA-12 | 1.94 | 13.3 | M | European | Normal | 6.3 | IVS1-13T>G/IVS1-13T>G | LOPD |
| GAA-13 | 1.97 | 13.5 | M | European | Normal | NP | IVS1-13T>G /p.G576S/p.E689K | Pseudo |
| GAA-14 | 2.11 | 14.5 | M | Asia | Normal | 12.7 | p.W746C/wt | Carrier PD |
| GAA-15 | 2.22 | 15.3 | F | European | Normal | NP | p.R178H/p.R178H | Pseudo |
| GAA-16 | 2.27 | 15.6 | F | South Asia | Normal | NP | IVS1-13T>G /p.G576S | Pseudo |
| GAA-17 | 2.52 | 17.3 | M | European | Normal | 0 | p.V222M/p.V222M | Pseudo |
| GAA-18 | 2.7 | 18.6 | M | European | Normal | NP | IVS1-13T>G /p.G576S/p.E689K | Pseudo |
| MPS I | NBS Enzyme Activity | % Enzyme Activity | Sex | Ethnic Origin | Urinary GAGs (mg/mmol creat) | Genotype | Predicted Phenotype |
|---|---|---|---|---|---|---|---|
| IDUA-01 | 0.1 | 0.9 | M | West Africa | NP | p.A79T/p.A79T | Pseudo |
| IDUA-02 | 0.17 | 1.6 | F | European | POS (DS 90.6 HS 220.6) | p.S16_A19del/p.Y201X | MPS I H |
| IDUA-03 | 0.2 | 1.9 | M | South Asia | NORMAL | NP | Pseudo |
| IDUA-04 | 0.22 | 2.1 | F | North Africa | POS (DS 121.7 HS 215.3) | p.P533R/p.P533R | MPS I H H/S |
| IDUA-05 | 0.38 | 3.6 | F | North Africa | NORMAL | p.R628G/p.R628G | VUS |
| IDUA-06 | 0.4 | 3.8 | M | West Africa | NORMAL | p.A79T/p.D223N | Pseudo |
| IDUA-07 | 0.41 | 3.9 | M | North Africa | NORMAL | p.A79T_ p.A361T/p.Y581X | Carrier/Pseudo |
| IDUA-08 | 0.49 | 4.6 | F | West Africa | NORMAL | p.A79T/p.A79T | Pseudo |
| IDUA-09 | 0.53 | 5.0 | F | West Africa | NP | p.A79T/p.D223N | Pseudo |
| IDUA-10 | 0.54 | 5.1 | M | West Africa | NP | p.A79T_p.T99I/p.D223N | Pseudo |
| IDUA-11 | 0.55 | 5.2 | F | European | NORMAL | p.L526P/p.L526P | VUS |
| IDUA-12 | 0.58 | 5.5 | F | West Africa | NP | p.A79T/p.A361T | Pseudo |
| IDUA-13 | 0.59 | 5.6 | F | North Africa | NORMAL | p.R263W/p.P650L | Pseudo |
| IDUA-14 | 0.66 | 6.3 | F | West Africa | NORMAL | p.A79T/p.A79T | Pseudo |
| IDUA-15 | 0.71 | 6.8 | F | European | NP | p.S16_A19del/p.H82Q | Carrier/Pseudo |
| IDUA-16 | 0.72 | 6.9 | M | West Africa | NORMAL | p.A79T/p.A79T | Pseudo |
| IDUA-17 | 0.85 | 8.1 | M | North Africa | NORMAL | p.A79T/p.R263W | Pseudo |
| IDUA-18 | 0.88 | 8.4 | M | NA | NORMAL | NP | Pseudo |
| IDUA-19 | 0.92 | 8.8 | F | West Africa | NORMAL | p.A79T/p.V322E | Pseudo |
| IDUA-20 | 0.97 | 9.3 | M | West Africa | NORMAL | p.A79T/p.F501L | Pseudo |
| IDUA-21 | 1.07 | 10.2 | M | NA | NORMAL | p.A79T/p.R263W | Pseudo |
| IDUA-22 | 1.1 | 10.5 | M | West Africa | NORMAL | p.A79T/p.S586F | Pseudo |
| IDUA-23 | 1.14 | 10.9 | F | West Africa | NORMAL | p.A79T/p.A79T | Pseudo |
| IDUA-24 | 1.16 | 11.1 | M | North Africa | NP | p.R263W/p.S586F | Pseudo |
| IDUA-25 | 1.26 | 12.0 | M | European | NORMAL | NP | Pseudo |
| IDUA-26 * | 2.21 | 21.1 | F | West Africa | NP | p.A79T/wt | Pseudo |
| Gaucher Disease | NBS Enzyme Activity | % Enzyme Activity | Sex | Ethnic Origin | Second Tier DBS LysoGb1 ** (nmol/L) | Genotype | Predicted Phenotype |
|---|---|---|---|---|---|---|---|
| ABG-01 | 0.44 | 4.5 | M | European | 163.8 | p.N409S/p.L483P | GD I |
| ABG-02 | 0.6 | 6.2 | M | European | 135.9 | p.N409S/p.N409S | GD I |
| ABG-03 | 0.88 | 9.0 | F | European | 77.4 | p.N409S/p.L483P | GD I |
| ABG-04 | 1.07 | 11.0 | M | European | 114.8 | p.N409S/p.N409S | GD I |
| ABG-05 | 1.07 | 11.0 | F | European | 116.6 | p.N409S/p.N227S | GD I |
| ABG-06 | 1.26 | 12.9 | M | European | 186.0 | p.N409S/Not found | Likely GD I |
| ABG-07 | 2.01 | 20.6 | M | European | 63.1 | p.N409S/p.N409S | GD I |
| Fabry Disease | NBS Enzyme Activity | % Enzyme Activity | Sex | Ethnic Origin | Second-Tier DBS LysoGb3 * (nmol/L) | Genotype | Predicted Phenotype [31] |
|---|---|---|---|---|---|---|---|
| GLA-01 | 0.64 | 6.1 | M | European | 1.02 | p.N215S | Type 2 FD |
| GLA-02 | 0.72 | 6.9 | M | North Africa | 1.79 | p.R363H | Type 2 FD |
| GLA-03 | 0.73 | 7.0 | M | European | 2.98 | p.R356G | Type 2 FD Likely |
| GLA-04 | 0.77 | 7.3 | M | East Asia | 0.75 | IVS4+919G>A | Type 2 FD |
| GLA-05 | 0.79 | 7.5 | M | European | 0.41 | p.M290L | Type 2 FD |
| GLA-06 | 0.87 | 8.3 | M | European | 0.73 | p.G116A | VUS (Not reported) |
| GLA-07 | 1.16 | 11.1 | M | East Asia | 0.62 | IVS4+919G>A | Type 2 FD |
| GLA-08 | 1.28 | 12.2 | M | European | 1.06 | p.L286V | VUS (Not reported) |
| GLA-09 | 1.37 | 13.1 | M | European | 0.83 | p.M51I | Type 2 FD |
| GLA-10 | 1.51 | 14.4 | M | West Africa | 0.96 | p.R356Q | Type 2 FD |
| GLA-11 | 2.05 | 19.6 | M | European | 0.54 | p.A143T | Benign |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. https://doi.org/10.3390/ijns5020024
Burlina AB, Polo G, Rubert L, Gueraldi D, Cazzorla C, Duro G, Salviati L, Burlina AP. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. International Journal of Neonatal Screening. 2019; 5(2):24. https://doi.org/10.3390/ijns5020024
Chicago/Turabian StyleBurlina, Alberto B., Giulia Polo, Laura Rubert, Daniela Gueraldi, Chiara Cazzorla, Giovanni Duro, Leonardo Salviati, and Alessandro P. Burlina. 2019. "Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy" International Journal of Neonatal Screening 5, no. 2: 24. https://doi.org/10.3390/ijns5020024
APA StyleBurlina, A. B., Polo, G., Rubert, L., Gueraldi, D., Cazzorla, C., Duro, G., Salviati, L., & Burlina, A. P. (2019). Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. International Journal of Neonatal Screening, 5(2), 24. https://doi.org/10.3390/ijns5020024