Congenital Hypothyroidism 3-Year Follow-Up Project: Region 4 Midwest Genetics Collaborative Results

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Study Design
3. Results
3.1. General
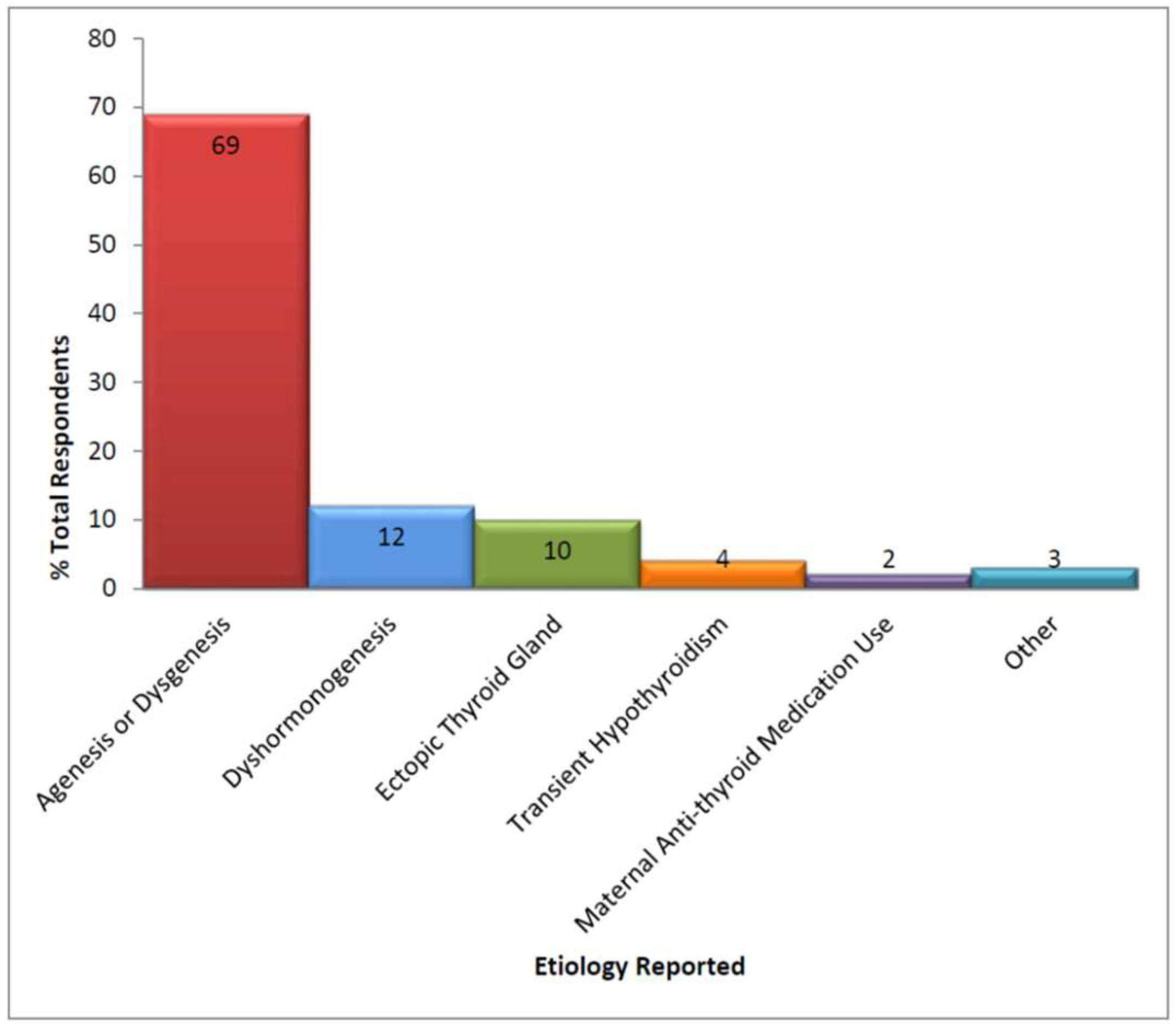
3.2. Diagnosis and Source of Care
3.3. Thyroid Management and Re-Evaluation
3.3.1. Clinician Responses
3.3.2. Parent Responses
3.4. Education and Genetic Counseling
3.4.1. Clinician Responses
3.4.2. Parent Responses
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- National Newborn Screening and Global Resource Center (NNSGRC). National Newborn Screening 2006 Incidence Report. 2006. Available online: http://genes-r-us.uthscsa.edu/sites/genes-r-us/files/resources/genetics/2006datareport.pdf (accessed on 12 June 2016).
- National Newborn Screening and Global Resource Center (NNSGRC). Conditions Screened by US Programs: National Newborn Screening Status Report. 2011. Available online: http://genes-r-us.uthscsa.edu/sites/genes-r-us/files/nbsdisorders.pdf (accessed on 12 June 2016).
- Rose, S.R.; Brown, R.S. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics 2006, 117, 2290–2303. [Google Scholar] [CrossRef] [PubMed]
- Léger, J.; Olivieri, A.; Donaldson, M.; Torresani, T.; Krude, H.; van Vliet, G.; Polak, M.; Butler, G.; ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE; Congenital Hypothyroidism Consensus Conference Group. European Society for Paediatric Endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J. Clin. Endocrinol. Metab. 2014, 99, 363–384. [Google Scholar] [CrossRef]
- Kemper, A.R.; Ouyang, L.; Grosse, S.D. Discontinuation of thyroid hormone treatment among children in the United States with congenital hypothyroidism: Findings from health insurance claims data. BMC Pediatr. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, S.J.; Grigorescu, V.; Kleyn, M.; Young, W.I.; Birbeck, G.; Todem, D.; Romero, R.; Paneth, N. Transient hypothyroidism at 3-year follow-up among cases of congenital hypothyroidism detected by newborn screening. J. Pediatr. 2013, 162, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wintergerst, K.; Gembel, G.; Kreipe, T.; Zeller, P.; Eugster, E.; Young, B.; Andruszewski, K.; Kleyn, M.; Cunningham, T.; Fawbush, S.; et al. Congenital Hypothyroidism Long-Term Follow-up Project: Navigating the Rough Waters of a Multi-Center, Multi-State Public Health Project. J. Genet. Couns. 2015, 24, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Szinnai, G. Genetics of normal and abnormal thyroid development in humans. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.L.; Davis, T.C.; Frempong, J.O.; Humiston, S.G.; Bocchini, A.; Kennen, E.M.; Lloyd-Puryear, M. Assessment of newborn screening parent education materials. Pediatrics 2006, 117 Pt 2, S320–S325. [Google Scholar] [CrossRef]
- Fant, K.; Clark, S.; Kemper, A. Completeness and complexity of information available to parents from newborn-screening programs. Pediatrics 2005, 115, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Varma, S.; Pediatric Endocrine Society. Educational Materials. Fact Sheets. 2018. Available online: https://www.pedsendo.org (accessed on 18 February 2018).


{kind=link}
| State | First-Tier Screening Method | Live Births | CH Diagnosis | Birth Prevalence |
|---|---|---|---|---|
| Michigan | TSH | 125,172 | 85 | 1:1473 |
| Minnesota | TSH | 73,675 | 46 | 1:1602 |
| Wisconsin | TSH | 72,757 | 43 | 1:1692 |
| Illinois | TSH | 180,530 | 94 | 1:1920 |
| Ohio | TSH | 150,784 | 75 | 1:2010 |
| Indiana | TSH | 89,719 | 42 | 1:2136 |
| Kentucky | T4/TSH | 58,507 | 24 | 1:2438 |
| Total | - | 751,144 | 409 | 1:1836 |
| Laboratory Studies | n | % | Imaging Studies | N | % |
|---|---|---|---|---|---|
| TSH | 162 | 97 | Thyroid ultrasound | 40 | 24 |
| Free thyroxine (T4) | 139 | 84 | Thyroid technetium scan | 50 | 29 |
| Total T4 | 28 | 16 | Thyroid uptake scan (I123) | 3 | 2 |
| Free triiodothyronine (T3) | 8 | 5 | Bone age X-ray | 4 | 2 |
| Total T3 | 5 | 3 | Brain MRI/CT | 1 | <1 |
| T3 Uptake | 1 | <1 | |||
| Thyroglobulin level | 4 | 2 | |||
| TBII/TSH receptor antibody | 4 | 3 | |||
| Anti-thyroid peroxidase and/or Anti-thyroglobulin antibodies | 3 | 2 | |||
| Maternal thyroid testing | 0 | -- |
| n = 138 | % | |
|---|---|---|
| Within 3 months | 134 | 97 |
| 2–4 weeks after discontinuation | 40 | 29 |
| 4–8 weeks after discontinuation | 83 | 60 |
| 8–12 weeks after discontinuation | 11 | 8 |
| Greater than 3 months | 4 | 3 |
| 3–6 months after discontinuation | 4 | 3 |
| 6–12 months after discontinuation | - |
| Source | CH Education * (n = 182) | Genetic Counseling * (n = 160) |
|---|---|---|
| n (valid %) | n (valid %) | |
| Face-to-face education in office | 170 (93) | 65 (41) |
| Printed literature | 105 (58) | 40 (25) |
| Internet references | 22 (12) | 3 (2) |
| Information sent by state agencies | 18 (10) | 1 (<1) |
| No standard form of education provided | 6 (3) | 72 (45) |
| Referral to pediatric endocrinologist | 3 (2) | 3 (2) |
| Referral to genetic specialist for counseling | - | 14 (9) |
| Source | Satisfied a (n = 48) | Not Satisfied a (n = 26) | χ2 |
|---|---|---|---|
| n (valid %) | n (valid %) | ||
| Pediatrician talked to parent | 30 (63) | 9 (35) | 5.26 * |
| Pediatric endocrinologist talked to parent | 42 (88) | 22 (85) | 0.12 |
| Genetic doctor/counselor talked to parent | 2 (4) | 1 (4) | <0.01 |
| Printed materials on CH | 29 (60) | 7 (27) | 7.57 ** |
| Internet sites were referred to me | 8 (17) | 4 (15) | 0.02 |
| Information received in the mail | 8 (17) | 2 (8) | 1.16 |
| I looked up information on my own via the Internet | 20 (42) | 11 (42) | <0.01 |
| No one explained the diagnosis or gave me information | 1 (1) |
| When to Re-Evaluate | When Re-Evaluation Can Be Waived |
|---|---|
| Definitive etiology not determined by diagnostic testing | Thyroid dysgenesis demonstrated by imaging (with exception of DUOX2 mutations or Pendred syndrome) |
| Initial treatment started for pre-term infants or during illness | Dyshormonogenesis confirmed by molecular genetic testing |
| Evidence of positive thyroid autoantibodies at diagnosis | TSH elevations above normal reference range for age after first year of life |
| No dosage increase in L-T4 needed since infancy as indicated by rise in TSH above normal range for age | |
| Negative molecular investigation for enzyme defect or whom no testing has been performed |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wintergerst, K.A.; Eugster, E.; Andruszewski, K.; Kleyn, M.; Vanderburg, N.; Sockalosky, J.; Menon, R.; Linard, S.; Kingery, S.; Rose, S.R.; et al. Congenital Hypothyroidism 3-Year Follow-Up Project: Region 4 Midwest Genetics Collaborative Results. Int. J. Neonatal Screen. 2018, 4, 18. https://doi.org/10.3390/ijns4020018
Wintergerst KA, Eugster E, Andruszewski K, Kleyn M, Vanderburg N, Sockalosky J, Menon R, Linard S, Kingery S, Rose SR, et al. Congenital Hypothyroidism 3-Year Follow-Up Project: Region 4 Midwest Genetics Collaborative Results. International Journal of Neonatal Screening. 2018; 4(2):18. https://doi.org/10.3390/ijns4020018
Chicago/Turabian StyleWintergerst, Kupper A., Erica Eugster, Karen Andruszewski, Mary Kleyn, Nancy Vanderburg, Joe Sockalosky, Ram Menon, Sharon Linard, Suzanne Kingery, Susan R. Rose, and et al. 2018. "Congenital Hypothyroidism 3-Year Follow-Up Project: Region 4 Midwest Genetics Collaborative Results" International Journal of Neonatal Screening 4, no. 2: 18. https://doi.org/10.3390/ijns4020018
APA StyleWintergerst, K. A., Eugster, E., Andruszewski, K., Kleyn, M., Vanderburg, N., Sockalosky, J., Menon, R., Linard, S., Kingery, S., Rose, S. R., Moore, J., Gembel, G., & Gorman, L. (2018). Congenital Hypothyroidism 3-Year Follow-Up Project: Region 4 Midwest Genetics Collaborative Results. International Journal of Neonatal Screening, 4(2), 18. https://doi.org/10.3390/ijns4020018