Are We Ready for Fragile X Newborn Screening Testing?—Lessons Learnt from a Feasibility Study

 ,
,
Abstract
:1. Introduction
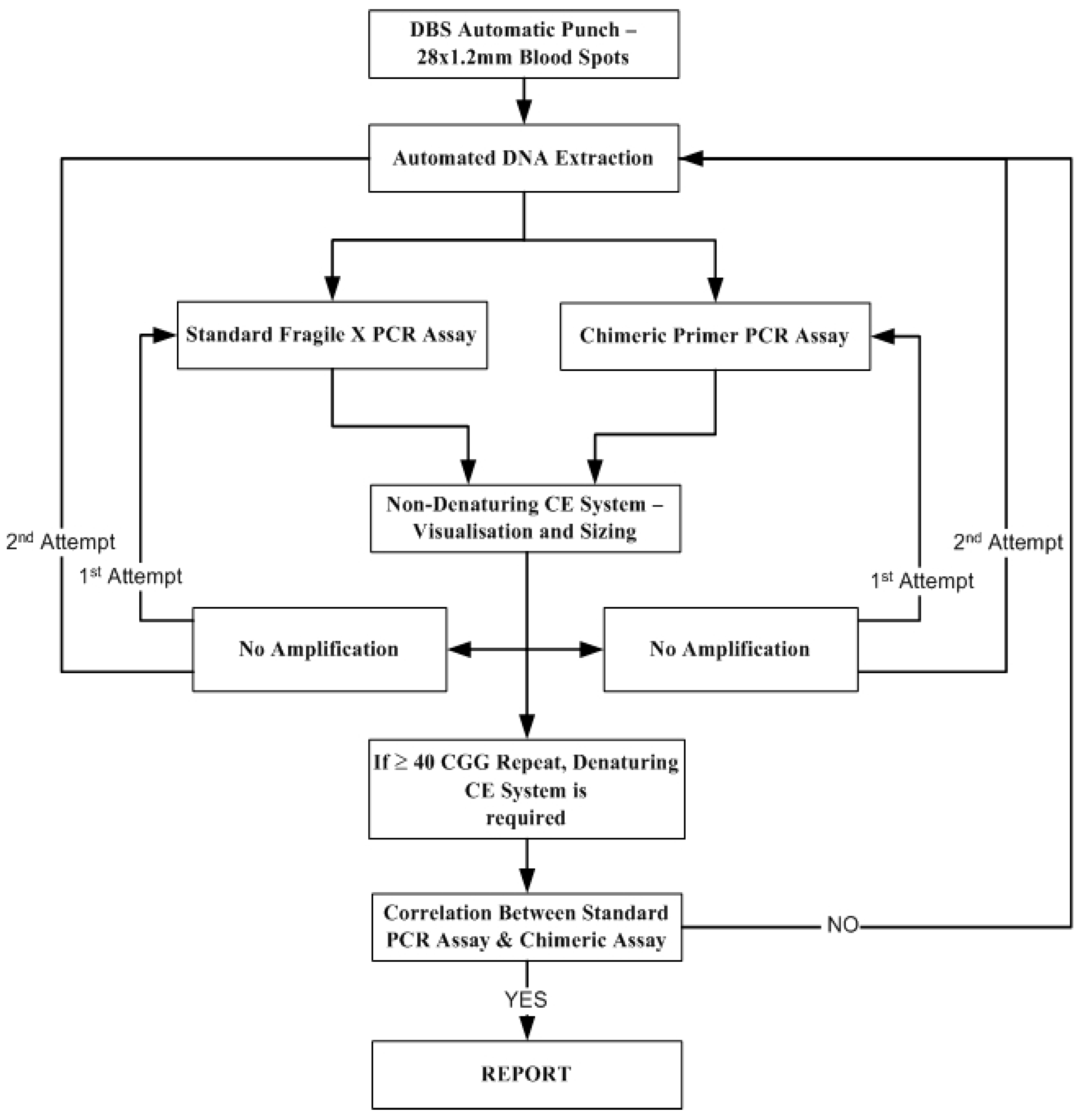
2. Materials and Methods
2.1. Sample Cohort for Study
2.2. DNA Extraction
2.3. Fragile X Fu PCR Assay
2.4. PCR Amplification for the Fragile X Method Using a Chimeric Primer
2.5. Sizing FMR1 Alleles
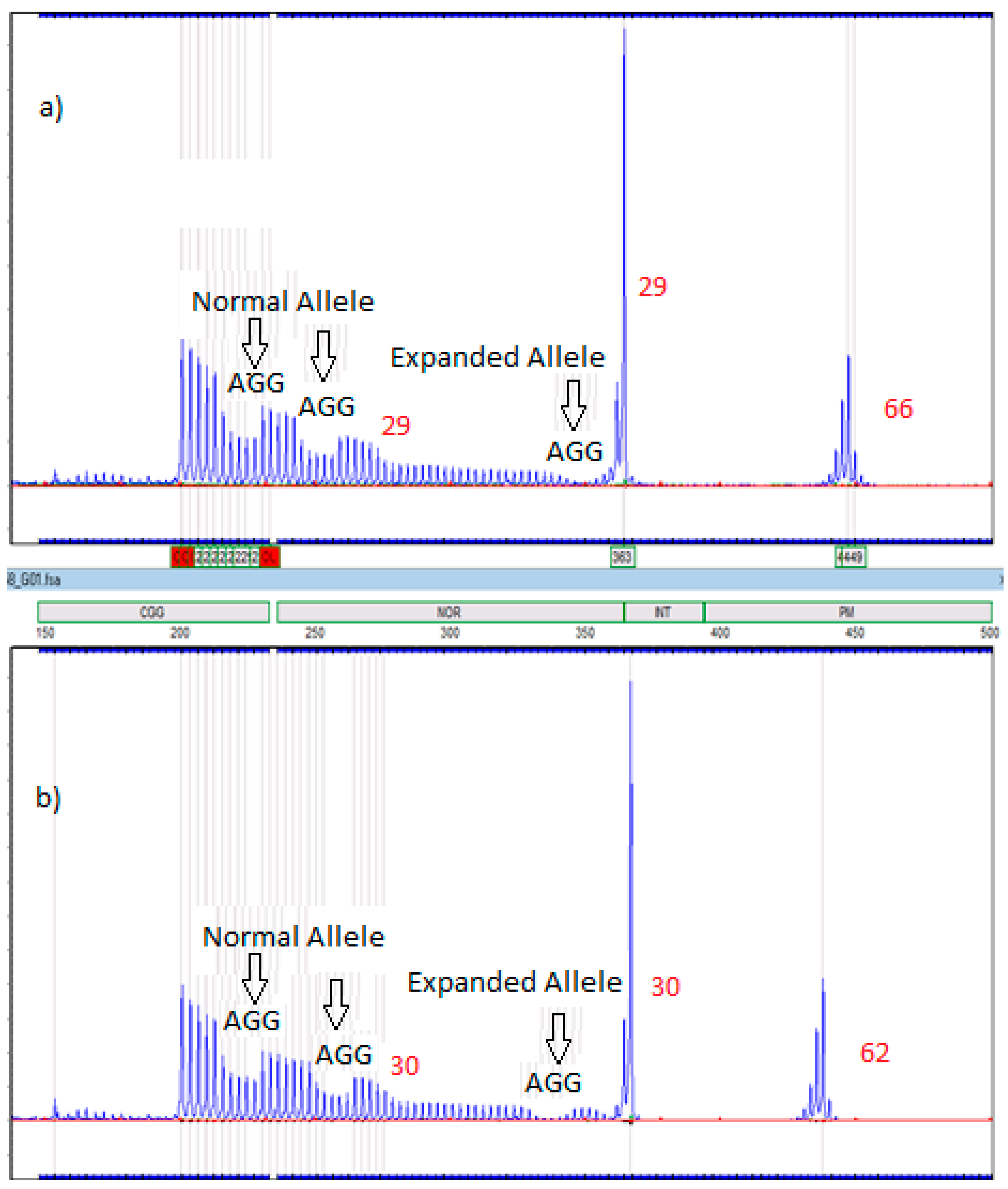
2.6. Amplidex Assay for AGG Interrupts
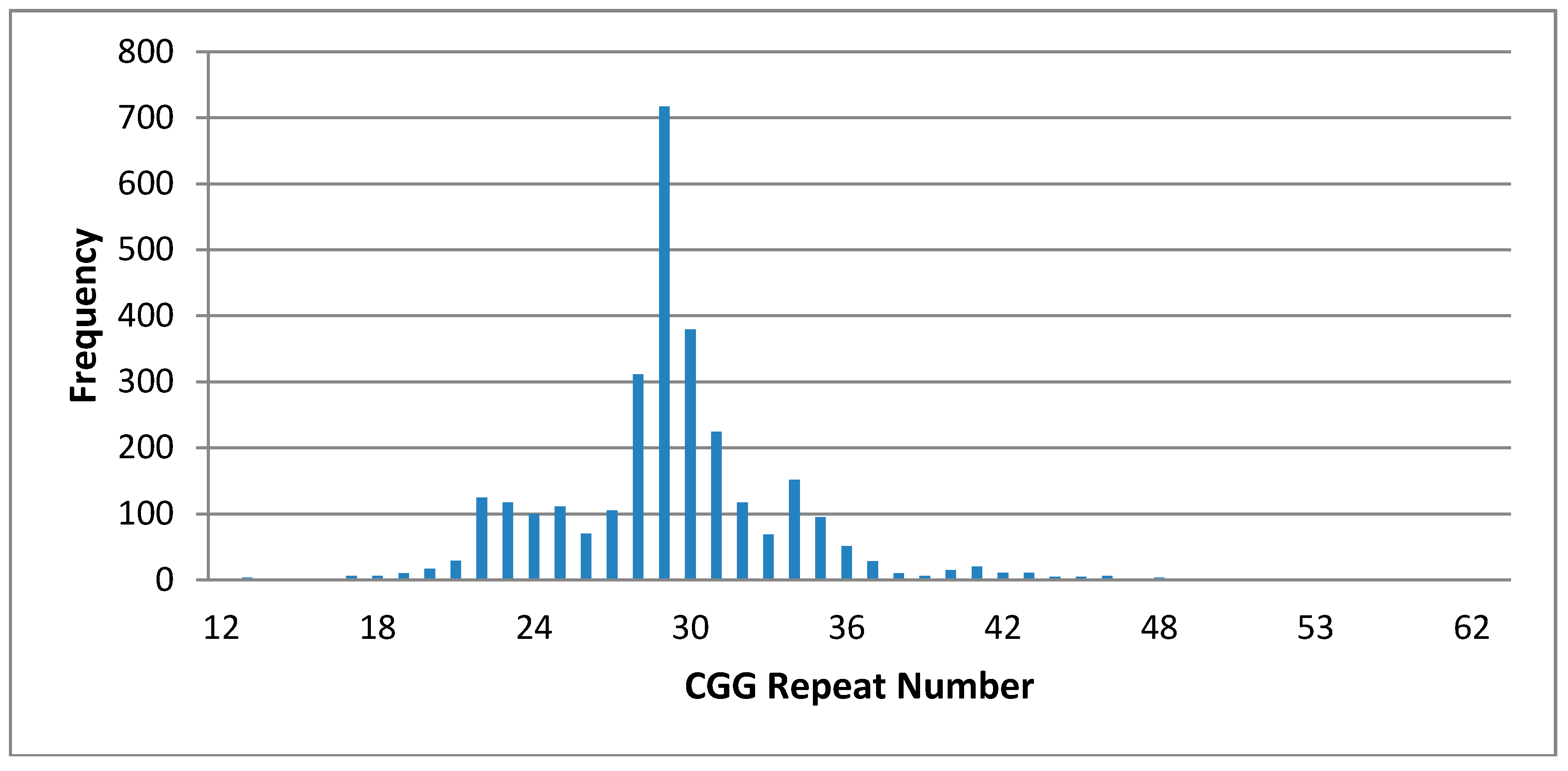
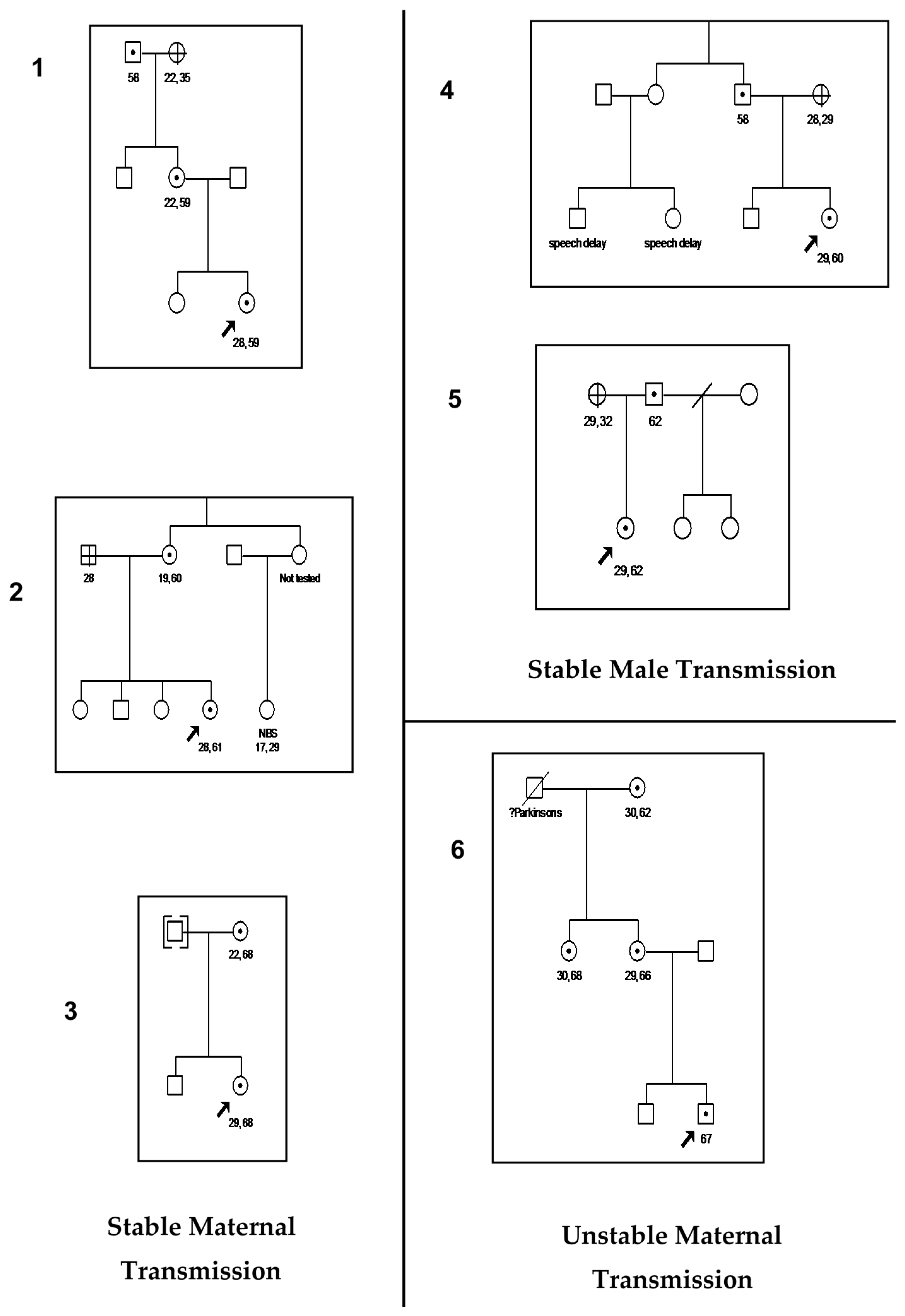
3. Results
Cost of Fragile X Chimeric Assay
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; Public health Paper Number 34; WHO: Geneva, Switzerland, 1968. [Google Scholar]
- Schaefer, G.B.; Lutz, R.E. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet. Med. 2006, 8, 549–556. [Google Scholar] [PubMed]
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. A 2014, 164A, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, F.R.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Booker, A.B.; Fallon, J.R. Regulating fragile X gene transcription in the brain and beyond. J. Cell. Physiol. 2005, 205, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Cuckle, H.; Taylor, G.; Hewison, J. Screening for fragile X syndrome. Health Technol. Assess. 1997, 1, 1–71. [Google Scholar]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, G.E., Jr.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Lopez Posadas, B.; Pan, R.; Raske, C.; Hagerman, P.J.; Tassone, F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Sah, S.; Glicksman, A.; Sherman, S.L.; Allen, E.; Berry-Kravis, E.; Tassone, F.; Yrigollen, C.; Cronister, A.; Jodah, M.; et al. Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am. J. Med. Genet. A 2013, 161, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V. Newborn screening methods for cystic fibrosis. Paediatr. Respir. Rev. 2003, 4, 272–277. [Google Scholar] [CrossRef]
- Borte, S.; von Döbeln, U.; Fasth, A.; Wang, N.; Janzi, M.; Winiarski, J.; Sack, U.; Pan-Hammarström, Q.; Borte, M.; Hammarström, L. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012, 119, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Holm, I.A.; Rehm, H.L.; McGuire, A.L.; Agrawal, P.B.; Parad, R.B.; Helm, M.H.; Genetti, C.A.; Beggs, A.H. The BabySeq Project: Preliminary Findings from a Randomized Trial of Exome Sequencing in Newborns. Presented at the American Society of Human Genetics 2016 Annual Meeting, Vancouver, BC, Canada, 18–22 October 2016. [Google Scholar]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.; Holden, J.J.; Fenwick, R.G.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A Rapid Polymerase Chain Reaction-Based Screening Method for Identification of All Expanded Alleles of the Fragile X (FMR1) Gene in Newborn and High-Risk Populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.; Gane, L.; Yarborough, M.; Hagerman, R.; Tassone, F. Newborn screening and cascade testing for FMR1 mutations. Am. J. Med. Genet. A 2013, 59–69. [Google Scholar] [CrossRef] [PubMed]
- The Fragile X Association of Australia Survey. Available online: https://fragilex.org.au/diagnosis-screening-for-fragile-x/ (accessed on 6 October 2015).
- Bailey, D.B., Jr.; Skinner, D.; Sparkman, K.L. Discovering fragile X syndrome: Family experiences and perceptions. Pediatrics 2003, 111, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Skinner, D.; Sparkman, K.L.; Bailey, D.B., Jr. Screening for Fragile X Syndrome: Parent attitudes and perspectives. Genet. Med. 2003, 5, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Osterweil, E.K.; Krueger, D.D.; Reinhold, K.; Bear, M.F. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar] [CrossRef] [PubMed]
- Çaku, A.; Pellerin, D.; Bouvier, P.; Riou, E.; Corbin, F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: An open-label study. Am. J. Med. Genet. A 2014, 164A, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.V.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 2009, 46, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Paribello, C.; Tao, L.; Folino, A.; Berry-Kravis, E.; Tranfaglia, M.; Ethell, I.M.; Ethell, D.W. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.J.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.; Wotton, T.; Bennetts, B.; Wiley, V.; Wilcken, B.; Rogers, C.; Boyle, J.; Turner, C.; Hansen, J.; Hunter, M.; et al. Maternal attitudes to newborn screening for fragile X syndrome. Am. J. Med. Genet. A 2013, 161, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Walichiewicz, P.; Xiaosen, X.; Pan, R.; Hagerman, P.J.; Tassone, F. Screening for Expanded Alleles of the FMR1 Gene in Blood Spots from Newborn Males in a Spanish Population. J. Mol. Diagn. 2009, 11, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Berry-Kravis, E.; Gane, L.W.; Guarda, S.; Hagerman, R.; Powell, C.M.; Tassone, F.; Wheeler, A. Fragile X Newborn Screening: Lessons Learned From a Multisite Screening Study. Pediatrics 2017, 139 (Suppl. 3), S216–S225. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, C.; Levesque, S.; Morel, M.L.; Rouillard, P.; Morgan, K.; Rousseau, F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: Loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum. Mol. Genet. 2002, 11, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Rouillard, P.; Morel, M.L.; Khandjian, E.W.; Morgan, K. Prevalence of carriers of premutation-size alleles of the FMRI gene--and implications for the population genetics of the fragile X syndrome. Am. J. Hum. Genet. 1995, 57, 1006–1018. [Google Scholar] [PubMed]
- Pesso, R.; Berkenstadt, M.; Cuckle, H.; Gak, E.; Peleg, L.; Frydman, M.; Barkai, G. Screening for fragile X syndrome in women of reproductive age. Prenat. Diagn. 2000, 20, 611–614. [Google Scholar] [CrossRef]
- Wang, Y.C.; Li, C.; Lin, M.L.; Lin, W.H.; Li, S.Y. Molecular diagnosis of fragile X syndrome and distribution of CGG repeats in the FMR-1 gene in Taiwanese. J. Formos. Med. Assoc. 2000, 99, 402–407. [Google Scholar] [PubMed]
- Robinson, H.; Wake, S.; Wright, F.; Laing, S.; Turner, G. Informed choice in fragile X syndrome and its effects on prevalence. Am. J. Med. Genet. 1996, 64, 198–202. [Google Scholar] [CrossRef]
- Turner, G.; Robinson, H.; Wake, S.; Laing, S.; Partington, M. Case finding for the fragile X syndrome and its consequences. BMJ 1997, 315, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B. Commentary on population screening for fragile X syndrome. Genet. Med. 2010, 12, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.K.; Archibald, A.D.; Cohen, J.; Metcalfe, S.A. A systematic review of population screening for fragile X syndrome. Genet. Med. 2010, 12, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Hayward, B.E.; Usdin, K. Improved Assays for AGG Interruptions in Fragile X Premutation Carriers. J. Mol. Diagn. 2017, 19, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, S.M.; Slater, H.R.; Francis, D.; Du Sart, D.; Li, X.; Amor, D.J.; Alliende, A.M.; Santa Maria, L.; Faundes, V.; Morales, P.; et al. Identification of Males with Cryptic Fragile X Alleles by Methylation-Specific Quantitative Melt Analysis. Clin. Chem. 2016, 62, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early detection of fragile X syndrome: Applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Lafauci, G.; Adayev, T.; Kascsak, R.; Kascsak, R.; Nolin, S.; Mehta, P.; Brown, W.T.; Dobkin, C. Fragile X Screening by Quantification of FMRP in Dried Blood Spots by a Luminex Immunoassay. J. Mol. Diagn. 2013, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cycles | Temperature | Time |
|---|---|---|
| 1 cycle | 98 °C | 10 min |
| 10 cycles | 97 °C | 45 s |
| 64 °C | 1 min | |
| 68 °C | 4 min | |
| 25 cycles | 97 °C | 35 s |
| 64 °C | 35 s | |
| 68 °C | 4 min (+20 s/cycle) | |
| 1 cycle | 68 °C | 10 min |
| Cycles | Temperature | Time |
|---|---|---|
| 1 cycle | 99 °C | 15 min |
| Hold (10 μL of PCR reaction mixture B added to each well) | 98 °C | |
| 1 cycle | 98 °C | 2 min |
| 10 cycles | 98 °C | 35 s |
| 64 °C | 35 s | |
| 68 °C | 4 min | |
| 27 cycles | 98 °C | 35 s |
| 64 °C | 35 s | |
| 68 °C | 4 min (+10 s/cycle) | |
| 1 cycle | 68 °C | 10 min |
| FX Allele CGG Range | Males n = 1013 | Females n = 987 |
|---|---|---|
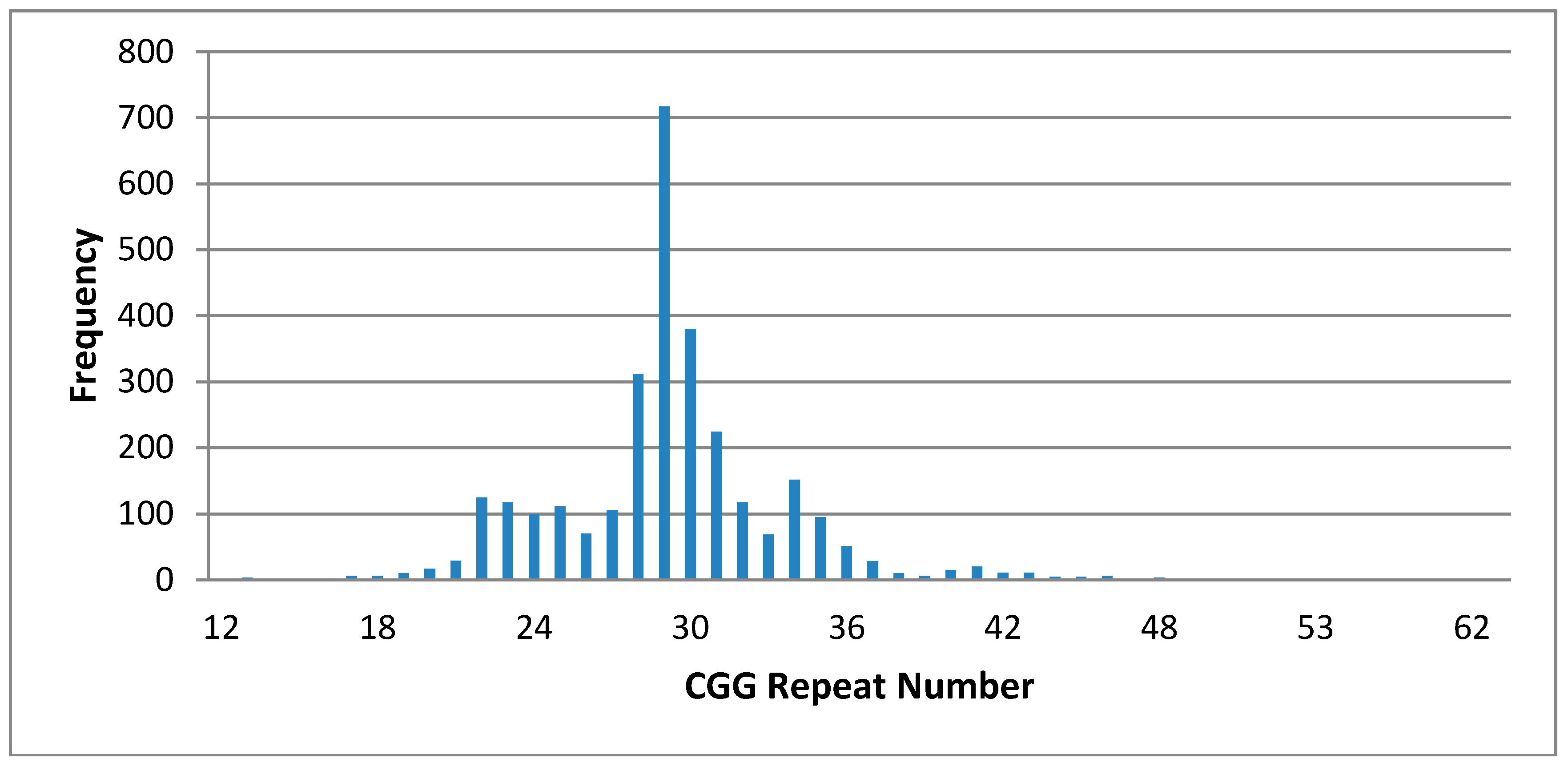
| 5–49 (n = 1986) | ||
| 40–44 | 17 | 42 |
| 45–49 | 6 | 11 |
| 50–54 | 1- 52 CGG | 3- 31, 51 CGG 29, 52 CGG 30, 53 CGG |
| 55–58 (Premutation: not reported) | 1- 55 CGG | 3- 29, 55 CGG 31, 56 CGG 29, 57 CGG |
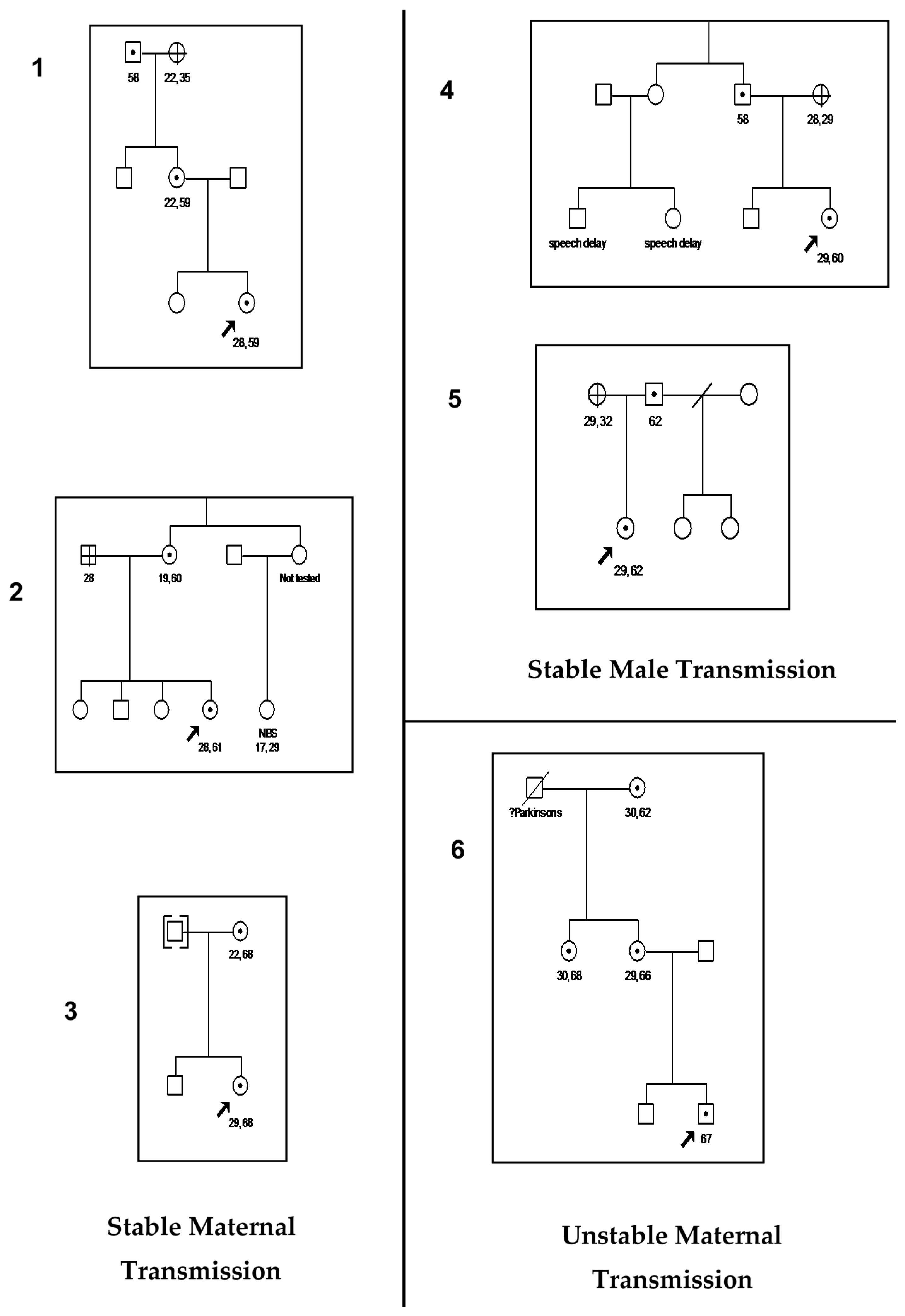
| 59–200 (Premutation: reported) | 1- 67 CGG * Unstable | 5- 28, 59 CGG * Stable 29, 60 CGG * Stable 28, 61CGG * Stable 29, 62 CCG * Stable 29, 68 CCG * Stable |
| >200 (Full Mutation) | Nil Found |
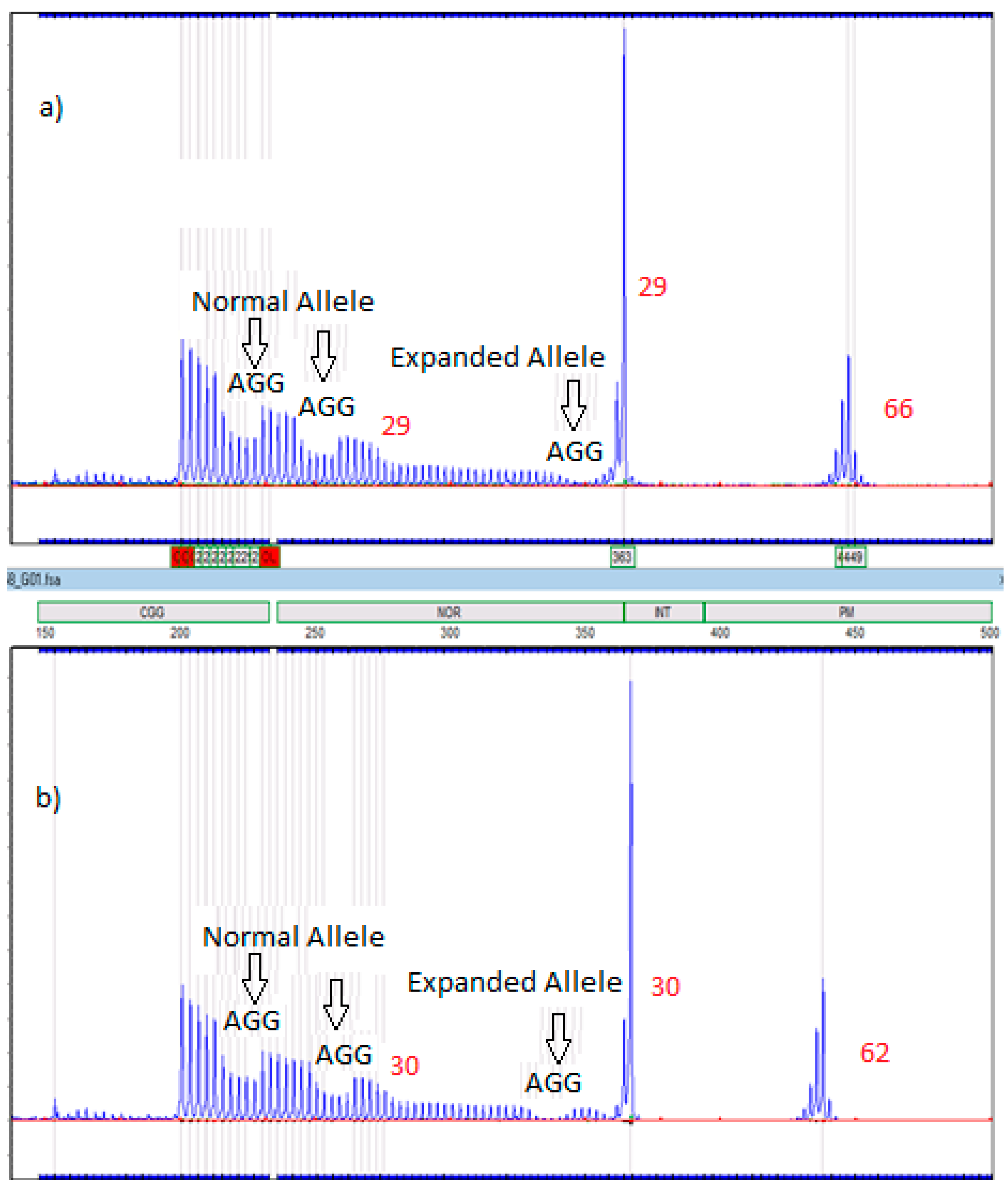
| Case Number | Family Member | AGG Number For Expanded Allele |
|---|---|---|
| 1 | Proband’s Mother | 2 |
| 2 | Proband’s Mother | 2 |
| 3 | Proband’s Mother | 3 |
| 4 | Proband’s Father | 1 |
| 5 | Proband’s Father | Insufficient sample |
| 6 | Proband’s Mother | 1 |
| Maternal Grandmother | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wotton, T.; Wiley, V.; Bennetts, B.; Christie, L.; Wilcken, B.; Jenkins, G.; Rogers, C.; Boyle, J.; Field, M. Are We Ready for Fragile X Newborn Screening Testing?—Lessons Learnt from a Feasibility Study. Int. J. Neonatal Screen. 2018, 4, 9. https://doi.org/10.3390/ijns4010009
Wotton T, Wiley V, Bennetts B, Christie L, Wilcken B, Jenkins G, Rogers C, Boyle J, Field M. Are We Ready for Fragile X Newborn Screening Testing?—Lessons Learnt from a Feasibility Study. International Journal of Neonatal Screening. 2018; 4(1):9. https://doi.org/10.3390/ijns4010009
Chicago/Turabian StyleWotton, Tiffany, Veronica Wiley, Bruce Bennetts, Louise Christie, Bridget Wilcken, Gemma Jenkins, Carolyn Rogers, Jackie Boyle, and Michael Field. 2018. "Are We Ready for Fragile X Newborn Screening Testing?—Lessons Learnt from a Feasibility Study" International Journal of Neonatal Screening 4, no. 1: 9. https://doi.org/10.3390/ijns4010009
APA StyleWotton, T., Wiley, V., Bennetts, B., Christie, L., Wilcken, B., Jenkins, G., Rogers, C., Boyle, J., & Field, M. (2018). Are We Ready for Fragile X Newborn Screening Testing?—Lessons Learnt from a Feasibility Study. International Journal of Neonatal Screening, 4(1), 9. https://doi.org/10.3390/ijns4010009