Duchenne Muscular Dystrophy Newborn Screening, a Case Study for Examining Ethical and Legal Issues for Pilots for Emerging Disorders: Considerations and Recommendations



{kind=link}
Abstract
:1. Background
2. Ethical and Legal Issues
- Universal screening (screening both males and females) for an X-linked disorder. In an X-linked disorder, the potential burdens and benefits are very different for males and females: DMD has variable expression in females, and carrier females have available preconception options, and
- The requirements imposed by present and proposed genotype-specific therapies in a condition where only a minority of identified patients would qualify for treatment.
3. Duchenne NBS Ethical and Legal Questions for Pilot Studies and Evidence Review
- How advocacy groups can best be engaged meaningfully, ensuring that patient perspectives are integrated into the evaluation and evidence review processes.
- How to design the parental permission process when the range of phenotypic variation associated with positive screens is high or when benefits and harms are not completely known.
- Consideration of issues surrounding universal versus gender-targeted screening for X-linked conditions.
- Assessment of benefit should include impact of minimizing the diagnostic odyssey for patient and family alike, and other salutary effects to the family including that of enabling non-medical interventions.
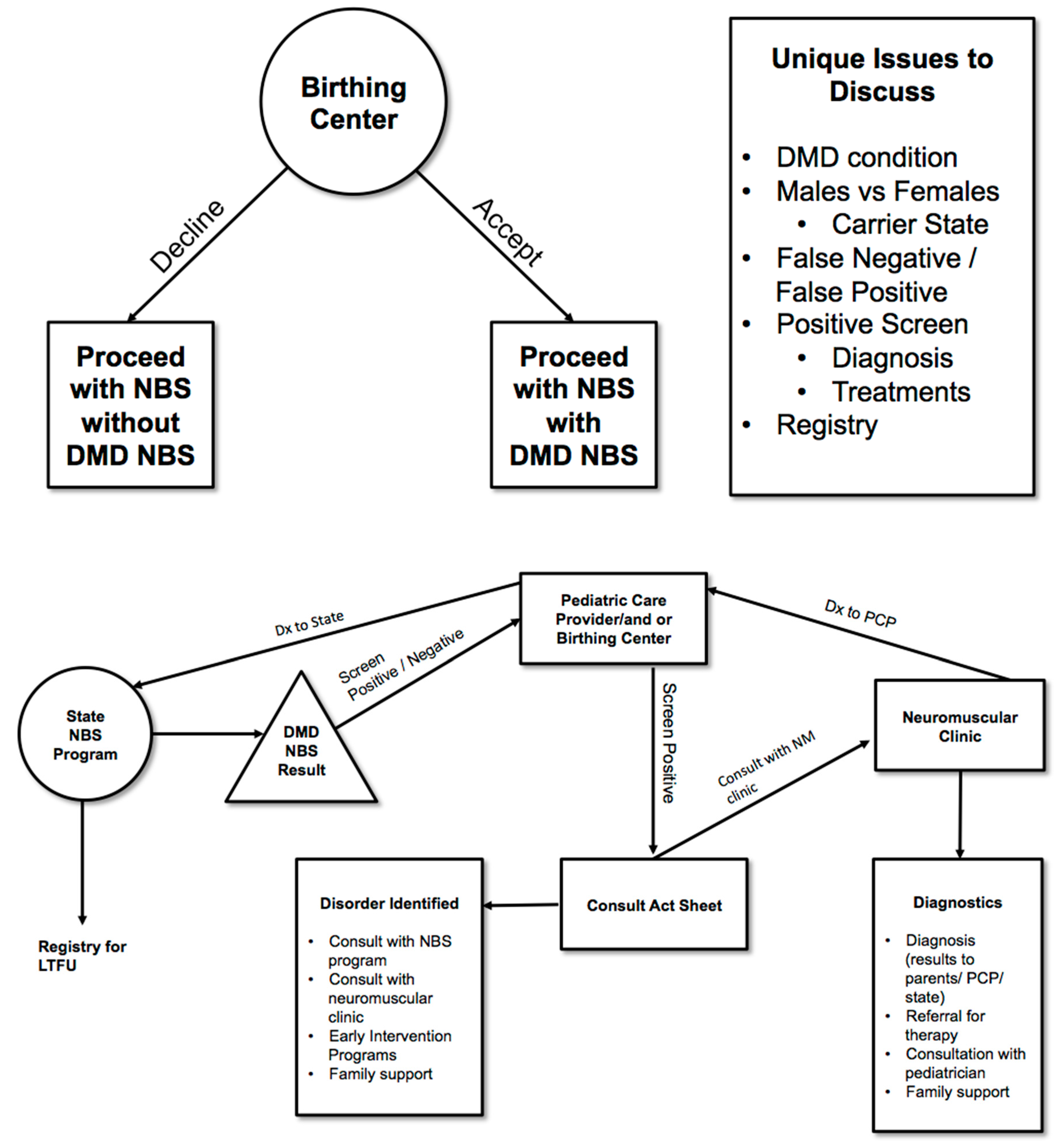
- Consideration of special difficulties that arise with identifying carrier status—Should Duchenne NBS include reporting of carrier status and if so then when, where and how to report? The Duchenne Workgroup recommended developing specific protocols to address these issues (See Figure 1).
- Consideration of incidental findings—Should Duchenne NBS include reporting of findings incidental to the screen or to the genetic analysis of the second tier testing and if so, when, where and how to report these findings? Incidental findings secondary to an elevated CK may include, for example, a dystrophy other than Duchenne. This secondary condition/incidental finding would be ruled out as Duchenne with subsequent genetic analysis and appropriate referral (See Figure 1).
- Role of industry in conducting pilots for newborn screening, whether for evaluating clinical devices for screening and diagnoses or for therapy and clinical trials—How does industry involvement in pilots affect pricing? How does industry involvement in pilots affect access to treatments post clinical trial? What are the ethical and legal issues if enrollment in a treatment trial is dependent on NBS?
4. Role of Advocacy Organizations in the NBS System
- Lay-advocates and researchers should continue to collaborate in research and policy development including the design of research studies.
- Efforts should be made to design patient-centered studies when structuring pilots or clinical trials. Pilots or clinical trial designs should not focus solely on the technical aspects of screening but the designs should include a broader range of issues to assure a patient-centered approach. Lay advocates, as with academic researchers, should disclose funding from drug companies or other entities that might contribute to the perception of bias.
- Lay-advocate leaders should guide their actions with transparency, clearly distinguishing between instances when they are speaking for themselves, for other lay-advocates or for their organization.
5. Considerations of Universal versus Targeted Screening and Reporting of Carriers and Incidental Findings
5.1. Question of Targeted or Universal Population Screening
- The Duchenne NBS pilot should be designed to conduct universal screening of newborns while concurrently exploring the many ethical and legal issues raised by screening for an X-linked disease. The issues include those raised in the above section as well as the following section addressing follow-up practices. An arm of the pilot might consider evaluation of parental attitudes on gender-targeted versus universal screening.
5.2. Follow-Up of Infrastructure for Families and Screened Positive Infants and How to Report Carrier Status and Incidental Findings
- Evaluate whether all infants identified with Duchenne or as carriers have received adequate follow-up including diagnosis after a positive screen, referral to appropriate clinical care centers, delivery of best practice treatment and management across the lifespan.
- Ensure that the partnering state NBS program have a short-term follow-up plan in place to ensure screen positive newborns receive proper referral to appropriate center for diagnosis and referral for treatments. This short-term follow-up program should:
- ▯
- Have diagnostic capacity in place that includes genetic sequencing, or referral for genetic sequencing, for screen positive infants.
- ▯
- Consider cascade genetic testing of both families and extended families. The project should have funds necessary to carry out appropriate genetic testing for diagnosis and in the family.
- ▯
- Include appropriate triage for infants with incidental findings such as sex chromosome disorders or other neuromuscular diseases.
- ▯
- Protocol for reporting of findings incidental to an elevated CK. The project should determine the appropriate CK (immunoassay) cut off to minimize false negatives and detection of carriers. A CK level needs to be determined that will identify all infants with Duchenne, and at the same time determine how frequently, if at all, female carriers are identified.
- ▯
- Determine an appropriate educational process that addresses public health and health care provider and parental genetic literacy about genotype/phenotype relationship/X-linked disorders/sex chromosome disorders.
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Al-Zaidy, S.A.; Lloyd-Puryear, M.A.; Kennedy, A.; Lopez, V.; Mendell, J.R. A roadmap to newborn screening for Duchenne Muscular Dystrophy. Int. J. Neonatal Screen. 2017, 3, 8. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, A.J.; Comeau, A.M.; Grosse, S.D.; Tanksley, S.; Prosser, L.A.; Ojodu, J.; Botkin, J.R.; Kemper, A.R.; Green, N.S. Evaluating harms in the assessment of net benefit: A framework for newborn screening condition review. Matern. Child Health J. 2016, 20, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Baily, M.A.; Murray, T.H. Ethics, evidence, and cost in newborn screening. Hastings Cent. Rep. 2008, 38, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Recommendations to HHS Secretary with Responses. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/recommendations-reports/index.html (accessed on 23 January 2018).
- Recommended Uniform Screening Panel. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html (accessed on 24 December 2017).
- The Advisory Committee on Heritable Disorders in Newborns and Children: Policies and Procedures for Operation and the Development of Recommendations for Screening Newborns and Children for Heritable Disorders and for the Heritable Disorders Program. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/about/index.html (accessed on 23 January 2018).
- Calonge, N.; Green, N.S.; Rinaldo, P.; Lloyd-Puryear, M.A.; Dougherty, D.; Boyle, C.; Watson, M.; Trotter, T.; Terry, S.F.; Howell, R.R. Committee Report: Method for evaluating conditions nominated for population-based screening of newborns and children. Genet. Med. 2010, 12, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.E.; Metternick-Jones, S.C.; Lister, K.J. International differences in the evaluation of conditions for newborn bloodspot screening: A review of scientific literature and policy documents. Eur. J. Hum. Genet. 2016, 25, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Burgard, P.; Hoffmann, G.F.; Lindner, M.; Loeber, J.G.; Rupp, K.; Taruscio, D.; Vittozzi, L. A framework to start the debate on neonatal screening policies in the EU: An Expert Opinion Document. Eur. J. Hum. Genet. 2014, 22, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Loeber, J.G.; Burgard, P.; Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Rupp, K.; Hoffmann, G.F.; Vittozzi, L. Newborn screening programmes in Europe; Arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J. Inherit. Metab. Dis. 2012, 35, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R. Newborn Screening: Toward a Uniform Screening Panel and System—Executive Summary. Pediatrics 2006, 117, S296–S307. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; World Health Organization: Geneva, Switzerland, 1968. [Google Scholar]
- Politano, L.; Nigro, V.; Nigro, G.; Petretta, V.R.; Passamano, L.; Papparella, S.; Di Somma, S.; Comi, L.I. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA 1996, 1, 1335–1338. [Google Scholar] [CrossRef]
- Therrell, B.L.; Lloyd-Puryear, M.A.; Mann, M.Y. Understanding newborn screening systems issues with emphasis on cystic fibrosis screening. J. Pediatr. 2005, 147, S6–S10. [Google Scholar] [CrossRef] [PubMed]
- Edwards, E.S.; Howell, R.R.; Lloyd-Puryear, M.A. Editors A look at newborn screening: Today and tomorrow. Pediatrics 2006, 117, S193–S354. [Google Scholar]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.; Apkon, S.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.; Alman, B.; Apkon, S.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.; Apkon, S.; Blackwell, A.; Colvin, M.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across lifespan. Lancet Neurol. 2018. [Google Scholar] [CrossRef]
- Barry, P.; Fryns, J.P.; Schotsmans, P.; Dierick, K. Carrier testing in minors: A systematic review of guidelines and position papers. Eur. J. Hum. Genet. 2006, 14, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Botkin, J.R.; Belmont, J.W.; Berg, J.S.; Berkman, B.E.; Bombard, Y.; Holm, I.A.; Levy, H.P.; Ormond, K.E.; Saal, H.M.; Spinner, N.B.; et al. Points to consider: Ethical legal and psychosocial implications of genetic testing in children and adolescents. Am. J. Hum. Genet. 2015, 97, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Report on the Genetic Testing of Children 2010; British Society for Human Genetics: Birmingham, UK, 2010.
- American Academy of Pediatrics/American College of Medical Genetics and Genomics. Technical Report: Ethical and policy issues in the genetic testing and screening of children. Genet. Med. 2013, 15, 234–245. [Google Scholar]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lloyd-Puryear, M.A.; Crawford, T.O.; Brower, A.; Stephenson, K.; Trotter, T.; Goldman, E.; Goldenberg, A.; Howell, R.R.; Kennedy, A.; Watson, M. Duchenne Muscular Dystrophy Newborn Screening, a Case Study for Examining Ethical and Legal Issues for Pilots for Emerging Disorders: Considerations and Recommendations. Int. J. Neonatal Screen. 2018, 4, 6. https://doi.org/10.3390/ijns4010006
Lloyd-Puryear MA, Crawford TO, Brower A, Stephenson K, Trotter T, Goldman E, Goldenberg A, Howell RR, Kennedy A, Watson M. Duchenne Muscular Dystrophy Newborn Screening, a Case Study for Examining Ethical and Legal Issues for Pilots for Emerging Disorders: Considerations and Recommendations. International Journal of Neonatal Screening. 2018; 4(1):6. https://doi.org/10.3390/ijns4010006
Chicago/Turabian StyleLloyd-Puryear, Michele A., Thomas O. Crawford, Amy Brower, Kristin Stephenson, Tracy Trotter, Edward Goldman, Aaron Goldenberg, R. Rodney Howell, Annie Kennedy, and Michael Watson. 2018. "Duchenne Muscular Dystrophy Newborn Screening, a Case Study for Examining Ethical and Legal Issues for Pilots for Emerging Disorders: Considerations and Recommendations" International Journal of Neonatal Screening 4, no. 1: 6. https://doi.org/10.3390/ijns4010006
APA StyleLloyd-Puryear, M. A., Crawford, T. O., Brower, A., Stephenson, K., Trotter, T., Goldman, E., Goldenberg, A., Howell, R. R., Kennedy, A., & Watson, M. (2018). Duchenne Muscular Dystrophy Newborn Screening, a Case Study for Examining Ethical and Legal Issues for Pilots for Emerging Disorders: Considerations and Recommendations. International Journal of Neonatal Screening, 4(1), 6. https://doi.org/10.3390/ijns4010006