
Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency


Abstract
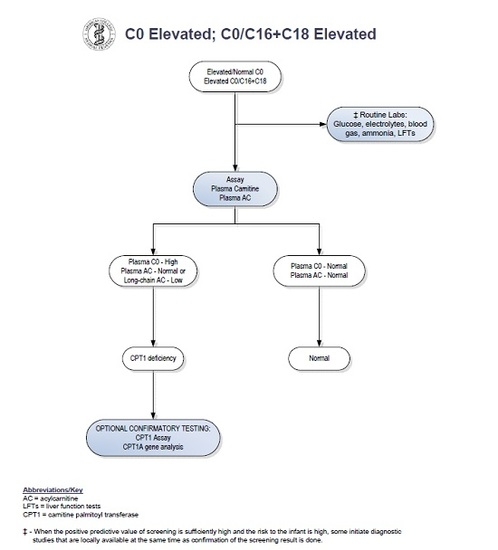
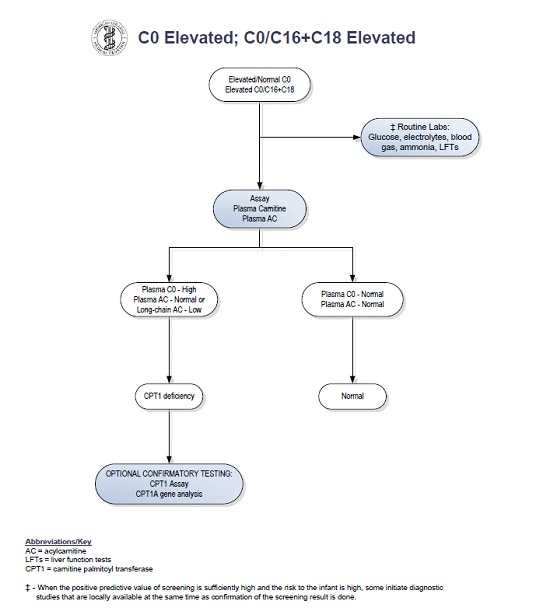
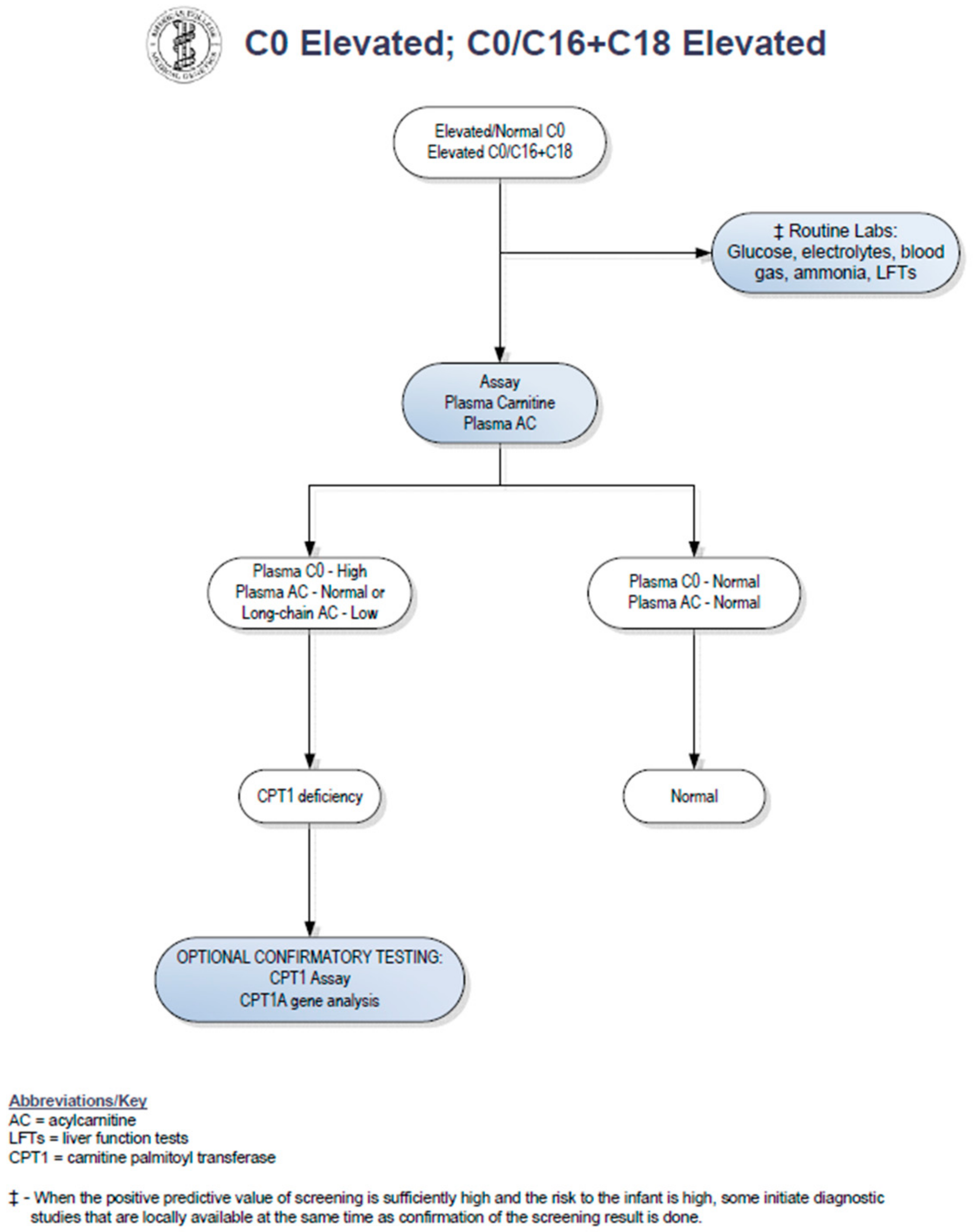
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
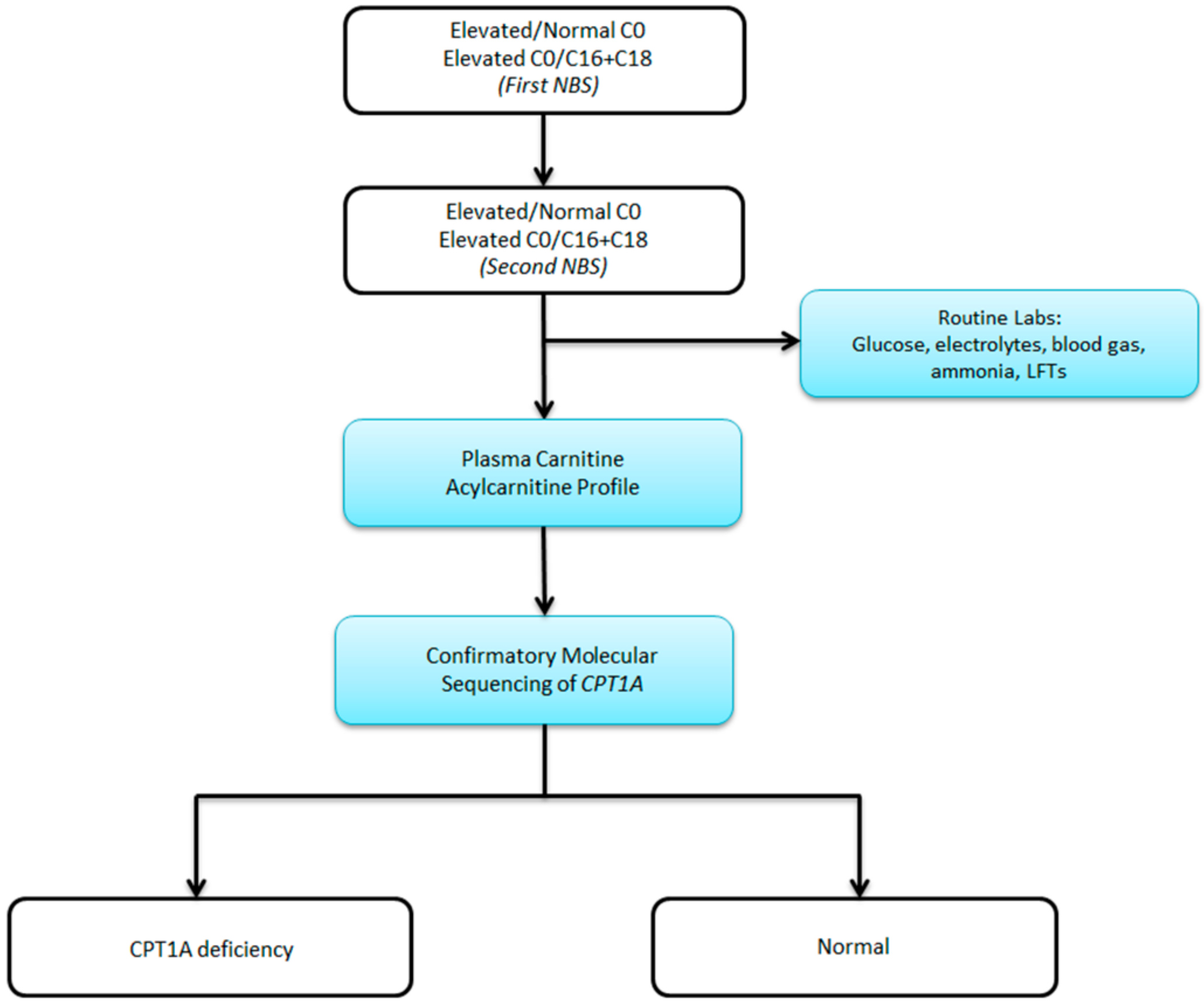
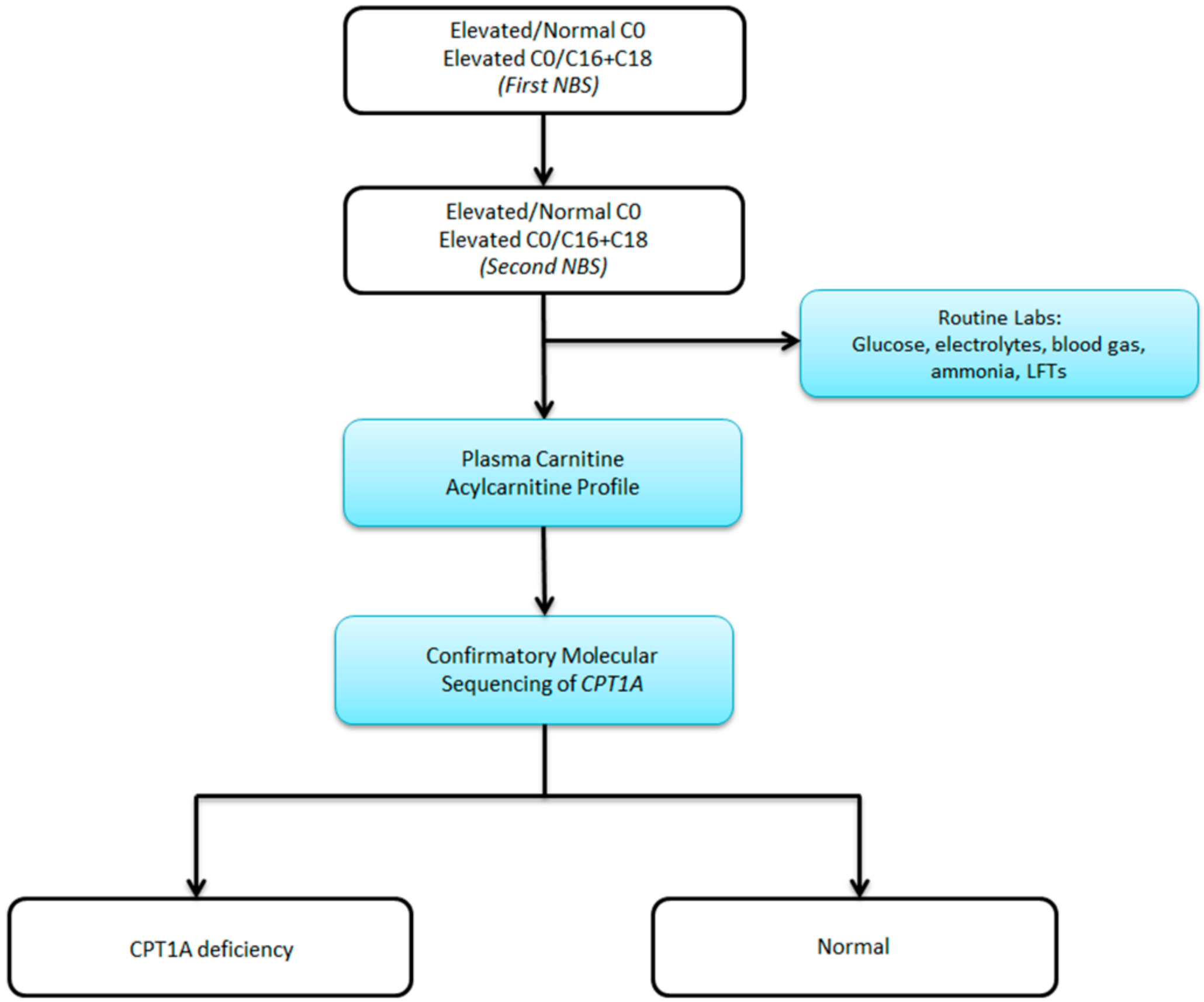
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Hughes, I. Pediatric Endocrinology and Inborn Errors of Metabolism, 1st ed.; Sarafoglou, K., Hoffman, G., Roth, K., Eds.; McGraw-Hill Education: New York, NY, USA, 2009. [Google Scholar]
- Clemente, F.J.; Cardona, A.; Inchley, C.E.; Peter, B.M.; Jacobs, G.; Pagani, L.; Kivisild, T. A selective sweep on a deleterious mutation in CPT1A in Arctic populations. Am. J. Human Gen. 2014, 95, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Korman, S.H.; Waterham, H.R.; Gutman, A.; Jakobs, C.; Wanders, R.J.A. Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency. Mol. Gen. Metab. 2005, 86, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Tsuburaya, R.; Sakamoto, O.; Arai, N.; Kobayashi, H.; Hasegawa, Y.; Yamaguchi, S.; Tsuchiya, S. Molecular analysis of a presymptomatic case of carnitine palmitoyl transferase I (CPTI) deficiency detected by tandem mass spectrometry newborn screening in Japan. Brain Dev. 2010, 32, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, R.; Roschinger, W.; Muntau, A.; Roscher, A. Hepatic carnitine palmitoyltransferase I deficiency: Acylcarnitine profiles in blood spots are highly specific. Clin. Chem. 2010, 47, 1763–1768. [Google Scholar]
- de Sain-van der Velden, M.; Verhoeven-Duif, N. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Gen. Metab. 2010, 110, 116–121. [Google Scholar] [CrossRef] [PubMed]
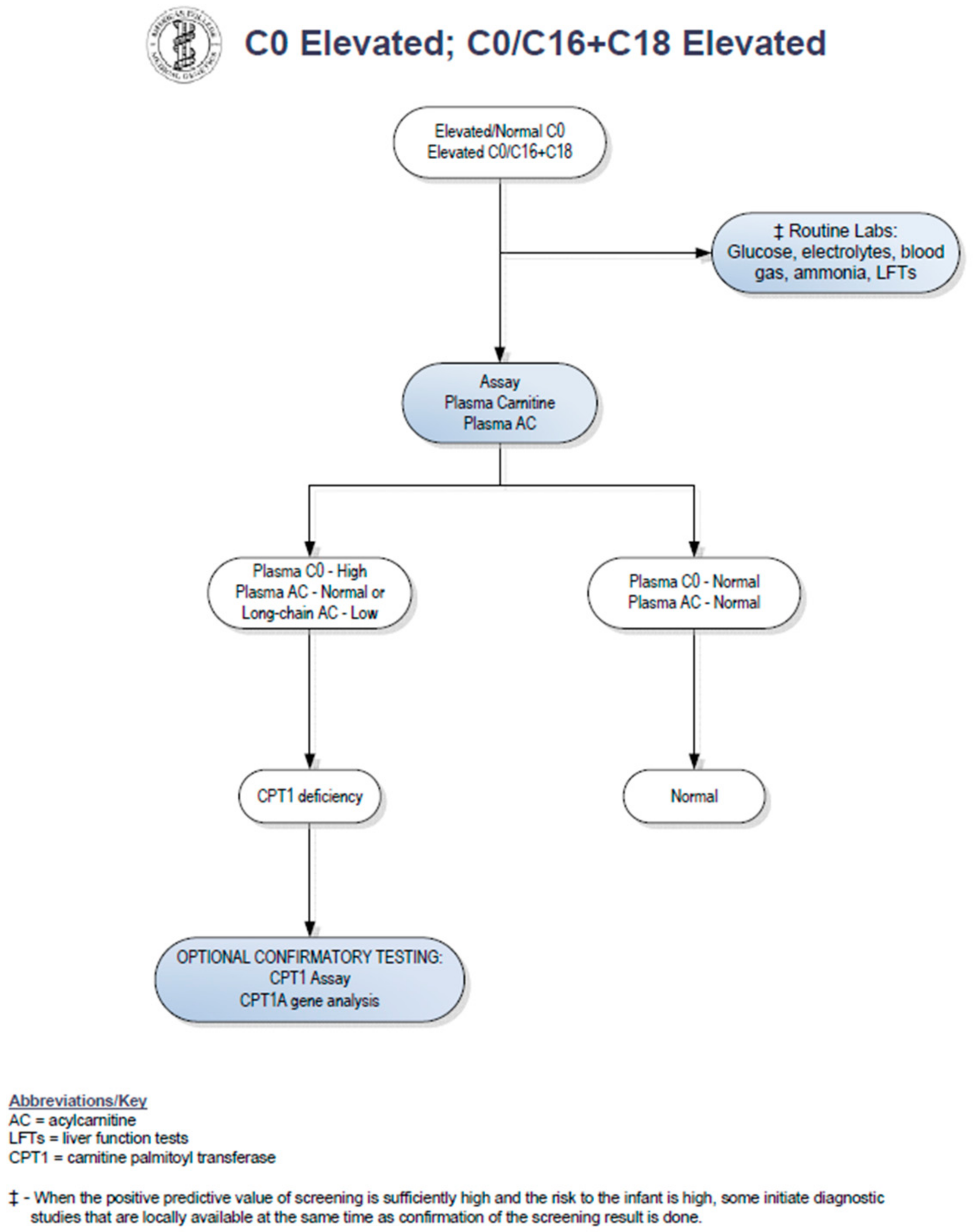
- American College of Medical Genetics and Genomics. Available online: http://www.acmg.net/StaticContent/ACT/Algorithms/Visio-C0.vsd;_C0.vsd;C16-C18.pdf.
- Stanley, C.; Palmieri, F.; Bennett, M. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID); Valle, D., Ed.; McGraw-Hill Education: New York, NY, USA, 2014. [Google Scholar]
- Edmondson, A.; Salant, J.; Ierardi-Curto, L.; Ficicioglu, C. Missed newborn screening case of Carnitine Palmitoyltransferase-II Deficiency. JIMD Rep. in press.
- Ficicioglu, C.; Coughlin, C.; Bennett, M.; Yudkoff, M. Very long-chain acyl-CoA dehydrogenase deficiency in a patient with normal newborn screening by tandem mass spectrometry. J. Pediatr. 2010, 156, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.; Vreken, P.; Ijlst, L. Disorders of mitochondrial fatty acyl-CoA β-oxidation. JIMD 1999, 22, 442–487. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowsett, L.; Lulis, L.; Ficicioglu, C.; Cuddapah, S. Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency. Int. J. Neonatal Screen. 2017, 3, 10. https://doi.org/10.3390/ijns3020010
Dowsett L, Lulis L, Ficicioglu C, Cuddapah S. Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency. International Journal of Neonatal Screening. 2017; 3(2):10. https://doi.org/10.3390/ijns3020010
Chicago/Turabian StyleDowsett, Leah, Lauren Lulis, Can Ficicioglu, and Sanmati Cuddapah. 2017. "Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency" International Journal of Neonatal Screening 3, no. 2: 10. https://doi.org/10.3390/ijns3020010
APA StyleDowsett, L., Lulis, L., Ficicioglu, C., & Cuddapah, S. (2017). Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency. International Journal of Neonatal Screening, 3(2), 10. https://doi.org/10.3390/ijns3020010