1. Introduction
Newborn screening has revolutionized the diagnosis of genetic and even non-genetic diagnosis. When I first began my medical career from medical school onward, the traditional way to diagnose inherited diseases, especially in children, was the presentation of problems, then history, physical examination, and some laboratory tests. Of course, early in my career, not many genetic disorders were known. However, the few that we did know—phenylketonuria (PKU) galactosemia, maple syrup urine disease (MSUD), congenital hypothyroidism, congenital adrenal hyperplasia—were always in infants or children already damaged with developmental delay or frank intellectual disability or severe liver disease, or even in a terminal condition ending in death. Today these and many other diagnoses are made through newborn screening, either within the first couple of days of life or at the first clinic visit within the first week or two, usually before any clinical features of the disorders appear. In other words, the diagnosis is an abnormal finding in the newborn screen, not from a set of clinical symptoms. One of the first things to which we expose physicians in our medical genetics training program is newborn screenings and the abnormal findings that can indicate a disorder. We then explain the features of the disorders as well as the confirmatory laboratory tests and the treatment.
This is wonderful news. It has dramatically altered the outcome of many metabolic disorders. It is rare to see a person with PKU detected in newborn screening who is intellectually disabled. When we want to illustrate intellectual disability from PKU in our training program, we have the trainees see a 65 or 70-year-old individual born before newborn screening who is brought in for care from a rehabilitation center or group home. Infants with MSUD or galactosemia no longer die. The course facies, lethargy and developmental delay that were once the hallmarks of congenital hypothyroidism have virtually disappeared as a result of newborn screening and early presymptomatic treatment with thyroid hormone replacement. Male infants with congenital adrenal hyperplasia, unlike the females who are born with their characteristic ambiguous genitalia that allows clinical diagnosis, no longer die of a salt-wasting crisis because the disease was not recognized at birth; they are detected by newborn screening and immediately treated.
Unfortunately, there is another side of newborn screening, unintended consequences, which is only now being given the attention it deserves. This side is the large number of newborn infants detected in newborn screening who have disorders or perhaps variants of serious disorders that are benign. This is not new to screening. It dates back to the beginning, when it was recognized that some identified babies with elevated phenylalanine in the screen had a much lower increase in phenylalanine and much higher tolerance for dietary phenylalanine therapies than other identified babies [
1,
2]. These infants were found to have a variant that we now know as benign mild hyperphenylalaninemia (MHP), not PKU, and they do not require a special diet. With expanded newborn screening by tandem mass spectrometry, likely benign findings have increased exponentially [
3]. Follow-up from expanded screening is beginning to show us that short chain acyl-CoA dehydrogenase deficiency (SCADD), a fatty acid oxidation disorder; and 3-methylcrotonyl-CoA carboxylase deficiency (3-MCCD), an organic acid disorder, are, at least, almost always certainly benign [
4,
5] and that perhaps the majority of infants detected with medium chain acyl-CoA dehydrogenase deficiency (MCADD) or very long chain acyl-CoA dehydrogenase deficiency (VLCADD), as well as fatty acid oxidation disorders, have mild variants of these two disorders that are also very likely benign [
6,
7]. How much harm do we do to families and the children identified by anxiety, unnecessary treatment and medical expenses, and unnecessarily “medicalizing” the child when we call attention to benign newborn screening findings? Hopefully, future study will allow us to sort the infants identified by newborn screening between those who have a true disease and require presymptomatic treatment and those with incidental benign “abnormalities” who require nothing and perhaps should not be identified.
I hope that the story of my journey in newborn screening will shed light on where I came from and how I came to appreciate the benefits of screening and the need to understand the full picture.
2. The Massachusetts Program: My Introduction to Newborn Screening
It was late Friday on a summer day in 1968. I was just finishing my 2-year fellowship in Metabolism at the Massachusetts General Hospital (MGH) in Boston. The phone rang in the Amino Acid Laboratory and I picked it up. Dr. Robert MacCready, the Director of the Biologic Laboratory of the Massachusetts Public Health Laboratories, who also directed the Massachusetts Newborn Screening Program, was calling, asking if anyone could come over to tell them what a “big amino acid spot” in a paper chromatogram could mean. At the time, newborn screening in Massachusetts had added this test to identify amino acid disorders not identified by the Guthrie bacterial assays for PKU, maple syrup urine disease, and galactosemia [
8]. I had never been to the State Laboratory Institute but I knew about newborn screening. In fact, it was the major reason I decided to go into metabolism rather than genetics (little did I know at that time that ‘metabolism’ would become ‘genetics’, at least the biochemical subdivision of genetics!). You see, two years before, as Chief Resident in Pediatrics at the Boston City Hospital, I had arranged for Dr. Mary Efron, a pioneer in the new field of metabolism and Director of the Amino Acid Laboratory at MGH to come over and give one of the talks for our monthly neonatology conferences. It was a terrible day in January, bitterly cold with six inches of snow on the ground. Only five showed up, one of whom was Dr. Efron. However, what we experienced was one of the most exciting talks I had ever heard. Dr. Efron described the studies she was conducting in identifying the metabolite abnormalities and enzymatic defects in several disorders, particularly hyperprolinemia and hydroxyprolinemia. However, more exciting was her description of newborn screening in Massachusetts and how, through presymptomatic detection and early treatment, it was changing the outcomes of PKU and other metabolic disorders. During my internship, I had been involved in the diagnosis of PKU in an 8-month-old boy who was admitted to the hospital for evaluation of developmental delay and seizures, so I knew how devastating PKU was (now 55 years later I still see John once a year). That newborn screening could prevent this from happening in PKU and other disorders blew my mind! When I heard the talk by Dr. Efron I had been accepted into the Genetics fellowship program at Children’s Hospital in Boston but after the talk I decided that what I really wanted to do was train in Metabolism. Subsequently I obtained a grant from the National Institutes of Health (NIH) and was accepted into Dr. Efron’s training program at MGH.
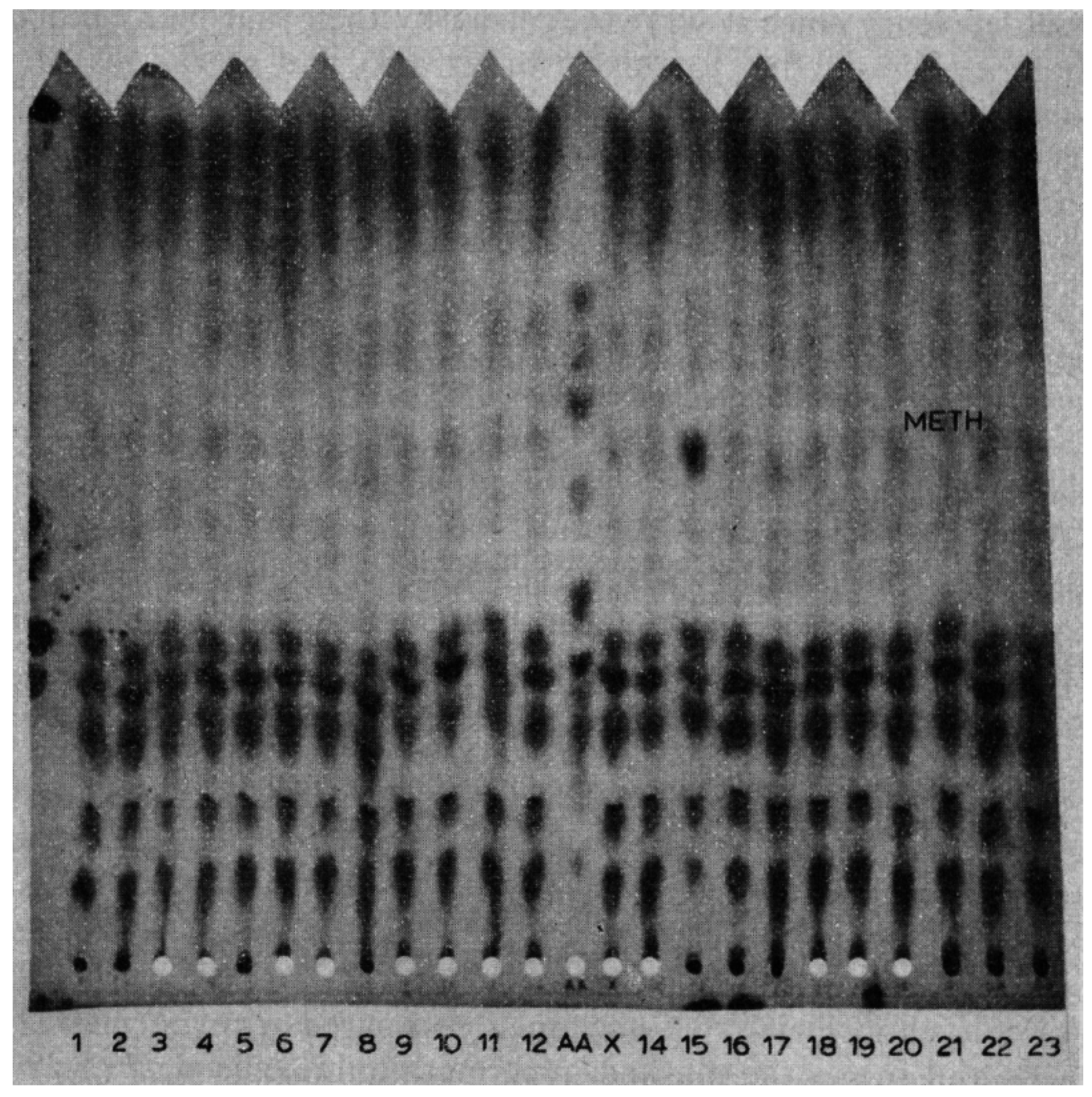
Back to the telephone call. I told Dr. MacCready I would be right over. I rode my bicycle (I was commuting by bike in those days) the 7 miles across town to the state laboratory where they showed me the chromatogram with the “spot” that I recognized as methionine (
Figure 1). I thought that most likely the baby had homocystinuria, the only disorder known at that time to be associated with elevated methionine. I subsequently saw the baby at the MGH and diagnosed homocystinuria in him as well as in his 2½-year-old sister who had severe developmental delay but without a diagnosis [
9]. The sister had never had metabolic testing and was born in Massachusetts before paper chromatography was added to the newborn screening. Interestingly, we recovered her original newborn blood specimen from storage and found that it also had hypermethioninemia on paper chromatography. This was my first experience with homocystinuria and it led to my lifelong interest in this and other disorders of methionine metabolism [
10,
11].
Upon seeing the process of newborn screening and realizing how vital this process was to the prevention of intellectual disability and other problems, I became hooked. With newborn screening, an entirely new dimension could be added to metabolic diseases and I wanted to be a part of this dimension. So, I began spending more time at the newborn screening laboratory, closely observing the performances of the bacterial assays and one-dimensional chromatography as well as the visualizing abnormalities that appeared from time to time. Shortly, the program began to depend upon me to interpret the abnormalities and connect with the medical community, i.e., having me call and advise the baby’s pediatrician as to how to proceed. When the abnormality was increased phenylalanine, I advised referral to the PKU clinic at Children’s Hospital. When it was any other abnormality, I advised referral to the MGH to be seen by myself or Dr. Vivian Shih. We very soon began having a unique experience with diagnosing and treating relatively large numbers of children with rare disorders.
In the second year, I served as consultant to the screening program. At the end of the year, I was asked if I might be interested in becoming Director of the screening program. I was interested but also wanted to continue pursuing my career in diagnosing and treating patients with metabolic disorders as well as the research in metabolic medicine that I was conducting at the MGH. By this time, I had completed my training fellowship and joined the active physician staff at the MGH in the Department of Neurology (the Joseph P. Kennedy, Jr. Memorial Laboratories for Mental Retardation under which the Amino Acid Laboratory resided, was established by the Kennedy family to be within the Department of Neurology at the MGH and the Harvard Medical School). So, I worked out an agreement to serve in the newborn screening program on a half-time basis with the title of Director of the Massachusetts Metabolic Disorders Program while staying at the MGH for the remainder of the time. This afforded the opportunity to continue following infants with non-PKU metabolic diseases in relation to their newborn screening findings. Later, in 1975, Massachusetts organized the New England Regional Newborn Screening Program that included Maine, Vermont, New Hampshire, and Rhode Island with Massachusetts as the screening laboratory. My title changed to Chief of Biochemical Genetics in the regional program but my metabolic activities remained the same. However, this expanded my view of metabolic disorders to beyond that limited to a single state.
3. My Introduction to Dr. Robert Guthrie
In the late 1960s, Dr. Robert Guthrie, the founder of newborn screening, organized an informal collaborative group composed of himself and members of his laboratory as well as representatives of Massachusetts, Oregon, New York (the Buffalo region) Ohio, and Maryland programs—the most active and comprehensive state newborn screening programs in the United States. The group met at least once a year. I was asked to represent Massachusetts, so I had the opportunity to observe his laboratory in Buffalo, to learn what was going on in other newborn screening programs, and to meet representatives of the federal agencies who were funding Dr. Guthrie, notably the Children’s Bureau of the U.S. Department of Public Health. At this time, Guthrie was interested in expanding newborn screening beyond PKU so as to include his bacterial inhibition assays for maple syrup urine disease and homocystinuria as well as the assay for galactosemia that he and Dr. Kenneth Paigen had together developed. The states represented in the collaborative group were selected for these additions but the focus of the collaborative group meetings was to develop a report of the first million babies screened for PKU (the report was never published).
The most important lesson I absorbed from these collaborative group meetings, though, was the woeful lack of medical input into newborn screening. Only Dr. Guthrie and I were physicians and I was the only one who had trained in metabolism and was actually diagnosing and treating patients. Soon I realized that the Massachusetts screening laboratory was unique in that it was the only state program in the country that had physicians within the laboratory (Dr. MacCready, myself, and, to a lesser extent, Dr. Shih). All other U.S. newborn screening laboratories were directed and operated by non-physicians. Moreover, in many of the laboratories the technicians were the responsible individuals, asked to make decisions about not only the operation of the laboratory and performance of the tests but to decide which tests and disorders to add to newborn screening. Dr. Guthrie and his laboratory had developed the bacterial assays for not only maple syrup urine disease, homocystinuria, and galactosemia but also for histidinemia and hyperlysinemia [
12]. He wanted all screening laboratories to add these tests. To do so, he had to rely primarily on the technicians in the laboratories or directors of maternal and child health programs who were not in the laboratories and had no training or experience in the metabolic disorders. I recognized this as an appalling situation since these decisions could affect the very lives of these babies, yet it was reality. Newborn screening was entirely separated from medicine. Not only did the screening laboratories have very limited understanding of the diseases being screened, but the medical community—primarily pediatric—had no idea how the screening was done or how the laboratories decided to report or not report an elevated metabolite or what beyond PKU was being screened (newborn screening was the “PKU test” and is still often referred to in that way), or even the location of the screening laboratory, and certainly had no knowledge of or experience in the disorders being detected, including PKU.
4. Connecting Newborn Screening to Medicine
During this time Dr. MacCready, Dr. Shih and I recognized the importance of investigating the diseases being identified and our unique position to perform these investigations as physicians intimately involved in newborn screening. Was PKU mostly a benign disorder, and were screening and early treatment not only unnecessary but actually harmful, as one notorious but influential physician asserted [
13]? Was histidinemia a “disease” or a benign disorder for which screening was contraindicated [
14]? What were our experiences in screening for galactosemia and were there problems in the screening procedures [
15]? Could PKU be detected by screening in the very early newborn specimen collected even before 24 hours of age [
16]? I became recognized as a national authority on the process of newborn screening and its relationship to the diagnosis and treatment of metabolic disorders. As a result, in 1975, Dr. Kurt Hirschhorn asked me to write a chapter on this subject for the
Advances in Human Genetics which he co-edited [
17]. This became a national and international reference and further consolidated my role as the medical contact for metabolic disorders to both the newborn screening and pediatric communities.
5. Newborn Screening and Children’s Hospital
One thing was missing in my metabolic activities—diagnosing, treating and following patients with PKU. This was a major omission since PKU was, as the great Charles Scriver described it, the “epitome of human biochemical genetics” [
18]. One day in 1978, I received a call from Dr. Mary Ellen Avery, Physician-in-Chief of the Department of Medicine at Boston Children’s Hospital, who asked if I would consider moving to Children’s Hospital as Director of the PKU Clinic and to expand the clinic to cover all of the metabolic disorders. Of course I was interested since it would provide the opportunity to treat and follow PKU. However, it provided something more, a support group composed of a nutritionist, social worker, psychologist, and other physicians. At the MGH, we had none of this, only myself and Dr. Shih. There was no social worker or psychologist to support the families and there was a different dietician every week, not someone who concentrated on metabolic disorders but a person from the hospital’s general dietetic staff. However, I was still conducting basic lab research at the MGH and I did not want to leave that nor my activities in the newborn screening program. In addition, the Children’s Hospital did not have a metabolic laboratory, only an amino acid analyzer, whereas at the MGH there was not only a dedicated amino acid analyzer but also gas chromatography—mass spectrometry for organic acid analysis, tissue culture capability, and other laboratory methodology.
Over the course of several months, I worked out a schedule that included all three entities—directing the now Metabolic Clinic at Children’s Hospital while continuing at the MGH and, very importantly, continuing my activities as Chief of Biochemical Genetics in the New England Regional Program. This triad continued for the next decade and resulted in a number of contributions to a further understanding of metabolic disorders and the role of newborn screening. Most importantly, Children’s Hospital began my lifetime collaboration with two outstanding individuals: Dr. Susan Waisbren, an extraordinary psychologist; and Frances (“Fran”) Rohr, an equally extraordinary nutritionist. Both have become national and international leaders in their respective metabolic fields. Moreover, the many other associations and collaborations with the outstanding faculty and students at Children’s Hospital and Harvard Medical School over the years have been of inestimable benefit to my professional career as well as a great joy.
6. Newborn Screening and Maternal PKU
One other very important direction resulted from my involvement with Children’s Hospital—maternal PKU. Actually, my introduction to maternal PKU began one day in the newborn screening program. Dr. MacCready told me about a newborn infant who had a transient phenylalanine elevation, i.e., upon repeat screening when the infant was a few days older, the phenylalanine level was normal. Dr. MacCready knew of this family and told me that this was maternal PKU, that the mother had PKU and the transient newborn elevations came from her. I had not heard about maternal PKU so I read the medical literature about such families and learned that the offspring from these pregnancies were believed to be microcephalic, to have congenital heart disease and intrauterine growth retardation (IUGR) and to become intellectually disabled. Dr. MacCready thought that obstetricians were not aware of this problem but should be made aware since he thought that identification of these pregnancies could lead to treatment that might prevent damage to the fetus. Consequently, he and I wrote an article for the
American Journal of Obstetrics and Gynecology in which we reported this family and reviewed the subject of maternal PKU [
19]. A few years later, Dr. Roger Lenke and I surveyed all of the unpublished and published cases of maternal PKU and published this information in what has become a classic reference on maternal PKU [
20]. At Children’s Hospital, Dr. Waisbren, Ms. Rohr and I, along with our colleagues in association with the newborn screening program, made maternal PKU a top priority. We established a New England Maternal PKU Program [
21], relied upon the results of an umbilical cord blood screening component of the newborn screening program to expand our understanding of maternal PKU [
22,
23], and subsequently assumed a major role in the international maternal PKU Collaborative Project that was developed and directed by Dr. Richard Koch [
24].
7. Tandem Mass Spectrometry and Further Expansion of Newborn Screening
From my very beginning in newborn screening, a primary interest was in expanding the coverage of metabolic disorders. I knew that beyond PKU there were many metabolic disorders similar to PKU in that they also caused developmental delay and often other abnormalities and might be effectively treated, but only if they, like PKU, were identified presymptomatically. This almost always meant newborn screening. In fact, my introduction to newborn screening was through homocystinuria, not PKU. My interest in expansion was greatly stimulated by the multiple bacterial assays of Guthrie, which had been added to PKU in the Massachusetts program, and the system of paper chromatography developed by Drs. Mary Efron and Hugo Moser that allowed for the visualization of all amino acids and was applied to filter paper specimens of blood and urine, also in the Massachusetts program [
8,
25]. Unfortunately, most of the disorders identified by paper chromatography were benign, so it had very limited value in the early diagnosis of important metabolic disorders [
26]. By the late-1970s and into the 1980s, newborn screening began to expand into medical areas beyond metabolic, specifically the early detection of the endocrine disorders such as congenital hypothyroidism and congenital adrenal hyperplasia as well as the hemoglobinopathies, primarily sickle cell disease [
27]. The sole metabolic exception was biotinidase deficiency, discovered and added to newborn screening by Dr. Barry Wolf in 1983–1984 [
28,
29]. In 1995, however, expansion of newborn screening returned to the metabolic disorders. Dr. David Millington and his group showed that tandem mass spectrometry (MS/MS) could be applied to the filter paper blood specimen used in newborn screening [
30] and Dr. Edwin Naylor showed, in a large private newborn screening program, that this methodology successfully identified important metabolic disorders not covered by standard newborn screening [
31]. However, the interpretation used by Naylor required a very laborious, time-consuming, manual examination of the print-out from each baby, and clearly not applicable to state newborn screening.
In 1996, a turning point occurred. At the meeting of the International Society for Neonatal Screening (ISNS) in Boston, Dr. Mohamed Rashed, working in Saudi Arabia, presented automated electrospray MS/MS that I realized could now allow MS/MS to be used for newborn screening of many metabolic disorders in all state and other centralized programs [
32]. I wanted this to be used in the Massachusetts program and hoped that it would quickly be extended to the New England Regional Newborn Screening Program. I was confident that MS/MS would not only greatly expand newborn screening for the metabolic disorders but was also a much more efficient and sensitive method for PKU and other metabolic screening that was being conducted by bacterial assays. Unfortunately, bureaucratic hindrance as well as legitimate disagreement led to a somewhat bitter dispute within the screening program and the Massachusetts Department of Public Health, which oversaw the program. I wrote an editorial in Clinical Chemistry advocating the addition of MS/MS to routine newborn screening and criticizing what I viewed as unreasonable obstruction to its implementation [
33]. I also acted as an advocate within the national Society of Inherited Metabolic Disorders (SIMD), which resulted in a strong statement supporting the addition of MS/MS. From this disagreement, however, came a significant advance—the formation of the Massachusetts Advisory Committee for Newborn Screening very ably led by Dr. Barry Zuckerman, Chairman of Pediatrics at the Boston Medical Center, and consisting of members of the newborn screening program and the Department of Public Health, representatives of the metabolic and genetic communities, lawyers, and parents [
34]. I was a member and served as a bridge between the newborn screening and metabolic communities. As it turned out, this Advisory Committee was part of a growing trend in all of the states, to place decisions about newborn screening in the hands of an omnibus advisory committee rather than arbitrarily leaving these decisions to a much more limited number of people or a single person. After much discussion, the Advisory Committee voted to have the Massachusetts program adopt MS/MS for one metabolic disorder—the fatty acid oxidation disorder known as medium chain acyl-CoA dehydrogenase deficiency (MCADD) which can cause sudden death—as well as for PKU and the other metabolic disorders that were screened by bacterial assays, and to allow its use for all of the additional metabolic disorders as well as cystic fibrosis on an optional basis with an opt-out provision [
35]. The controversy, however, eventually led to a rather uncomfortable relationship and eventually to my resignation from the New England Regional Newborn Screening Program.
It was also time for me to now concentrate my professional career in one place, Boston Children’s Hospital, where I could devote myself to patient care and clinical research. Nevertheless, I owed and continue to owe a great debt of gratitude to newborn screening in Massachusetts for providing me with so much that has been rich and rewarding. Then, as now, newborn screening has not only been a major interest but also an extraordinary part of my professional life.


{kind=link}