
Brain Biomarkers of Long-Term Outcome of Neonatal Onset Urea Cycle Disorder


Abstract
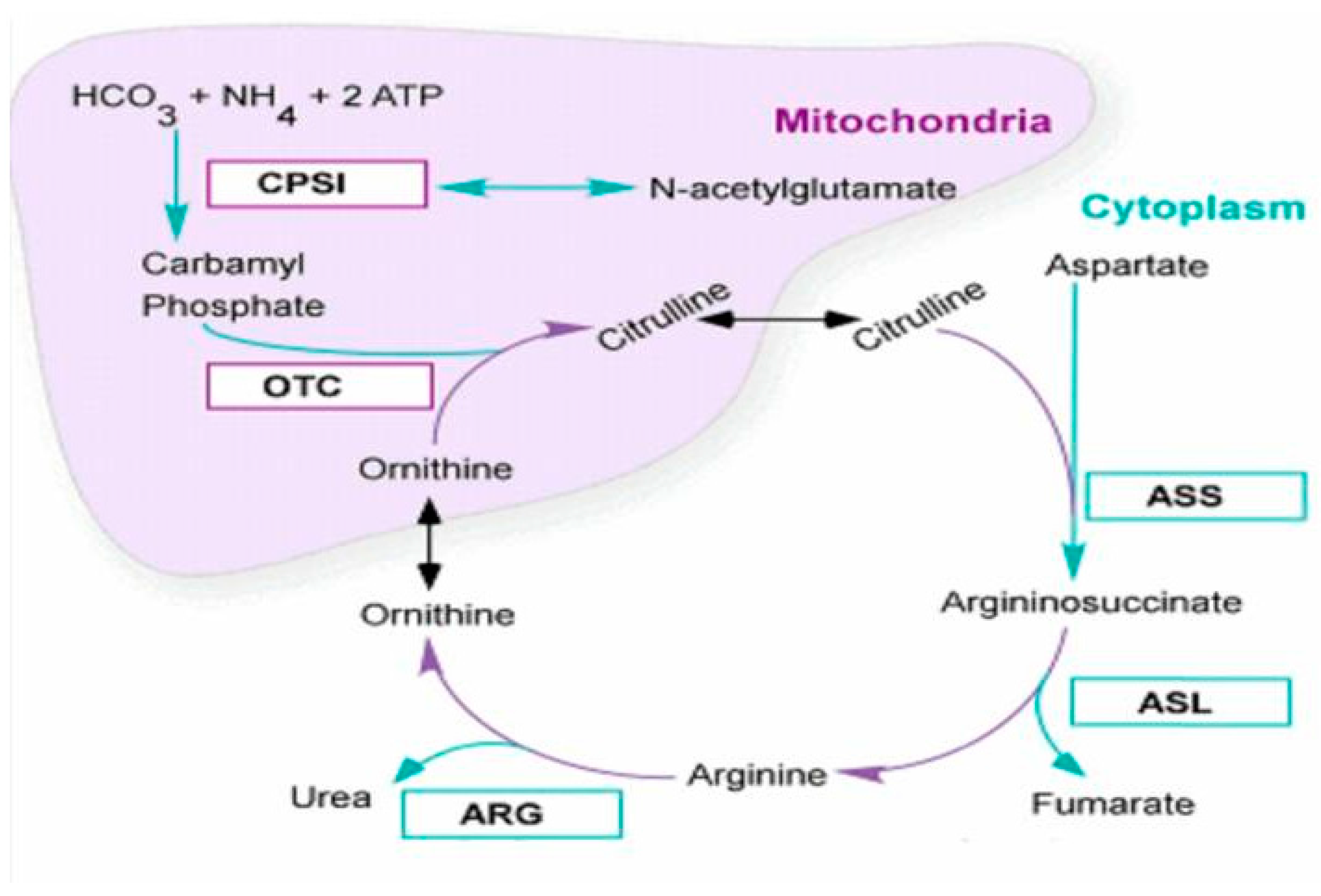
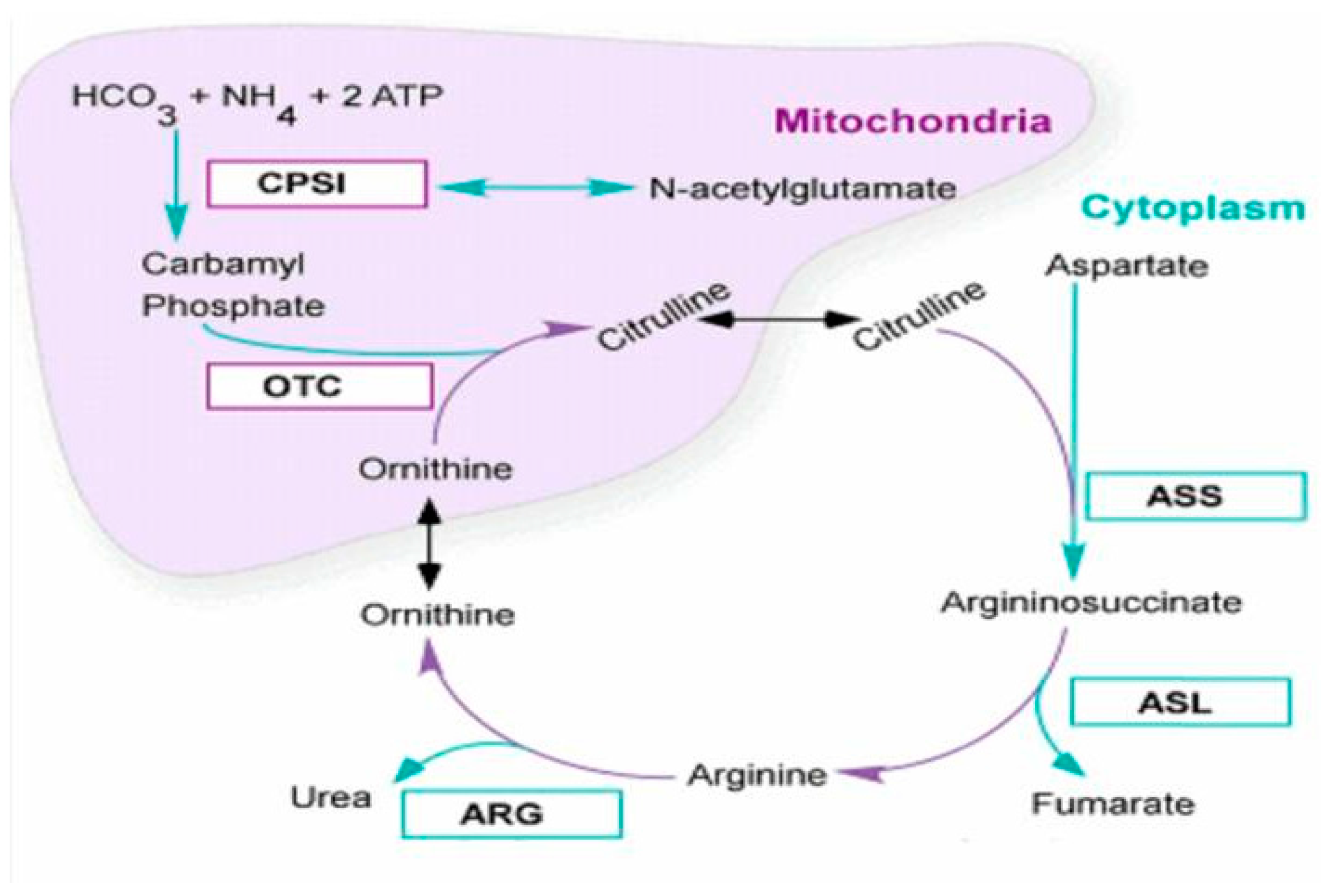
:1. Introduction
2. Patient and Methods
2.1. Presentation of the Case
2.2. Neuroimaging Collection
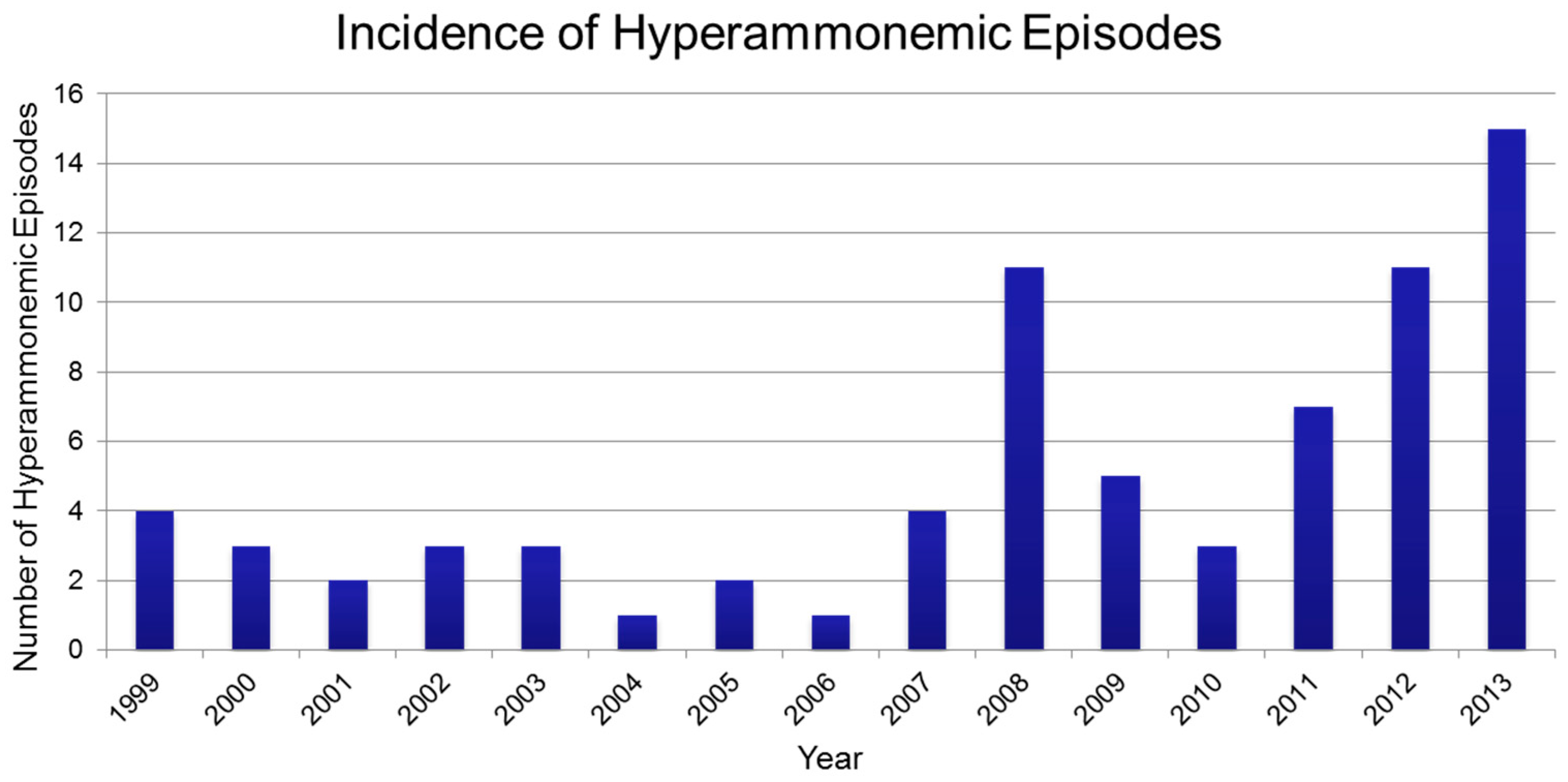
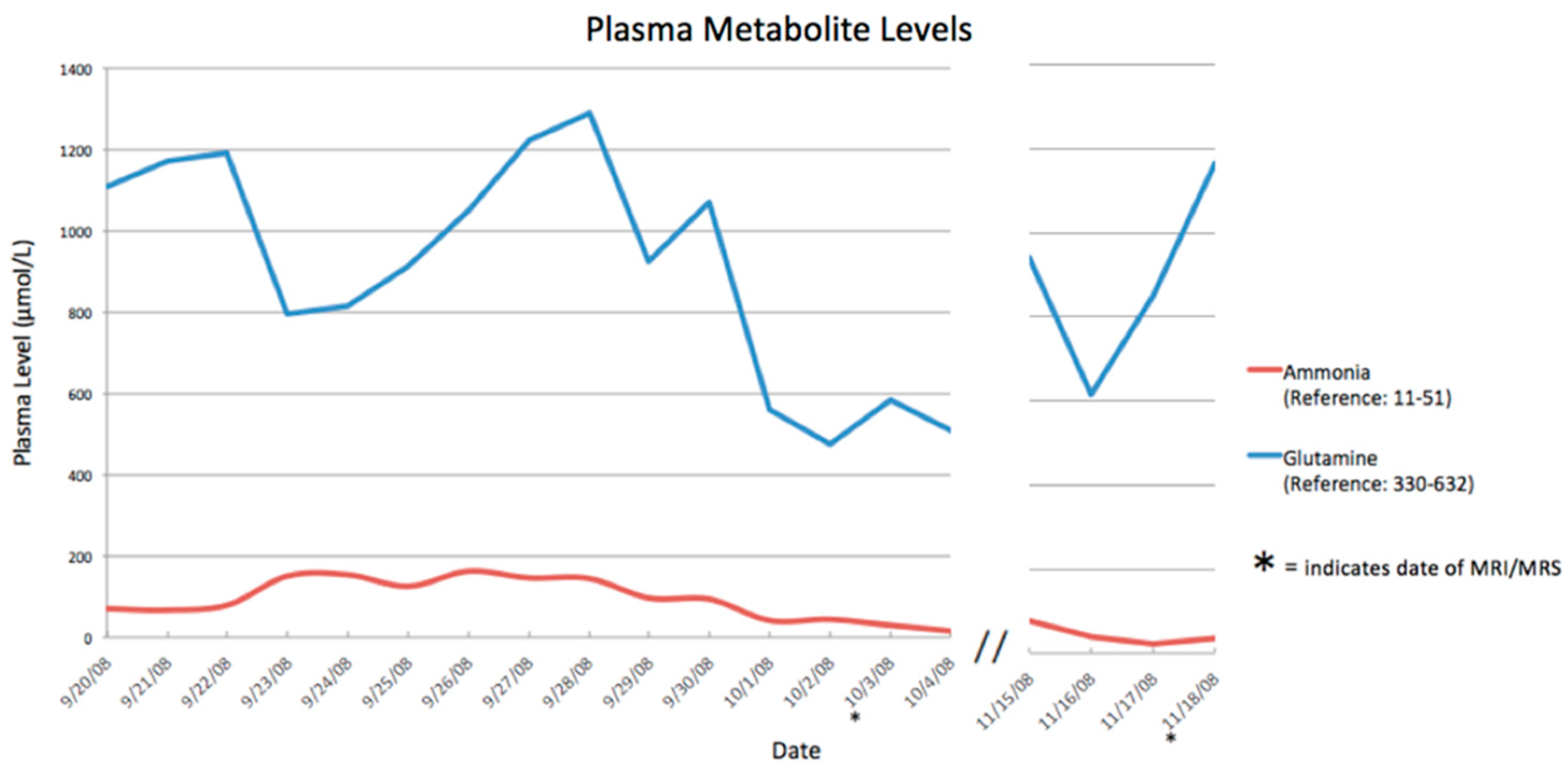
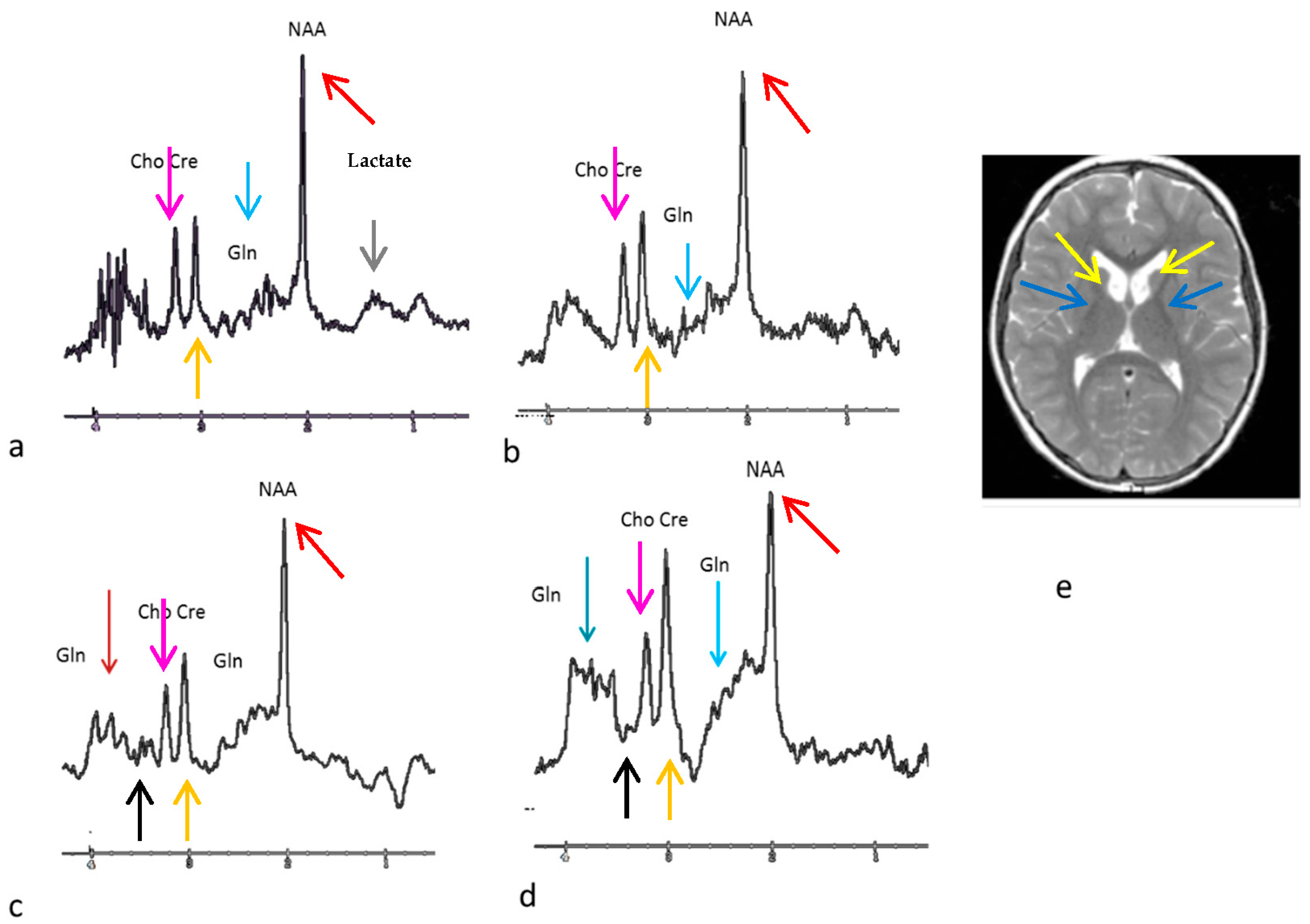
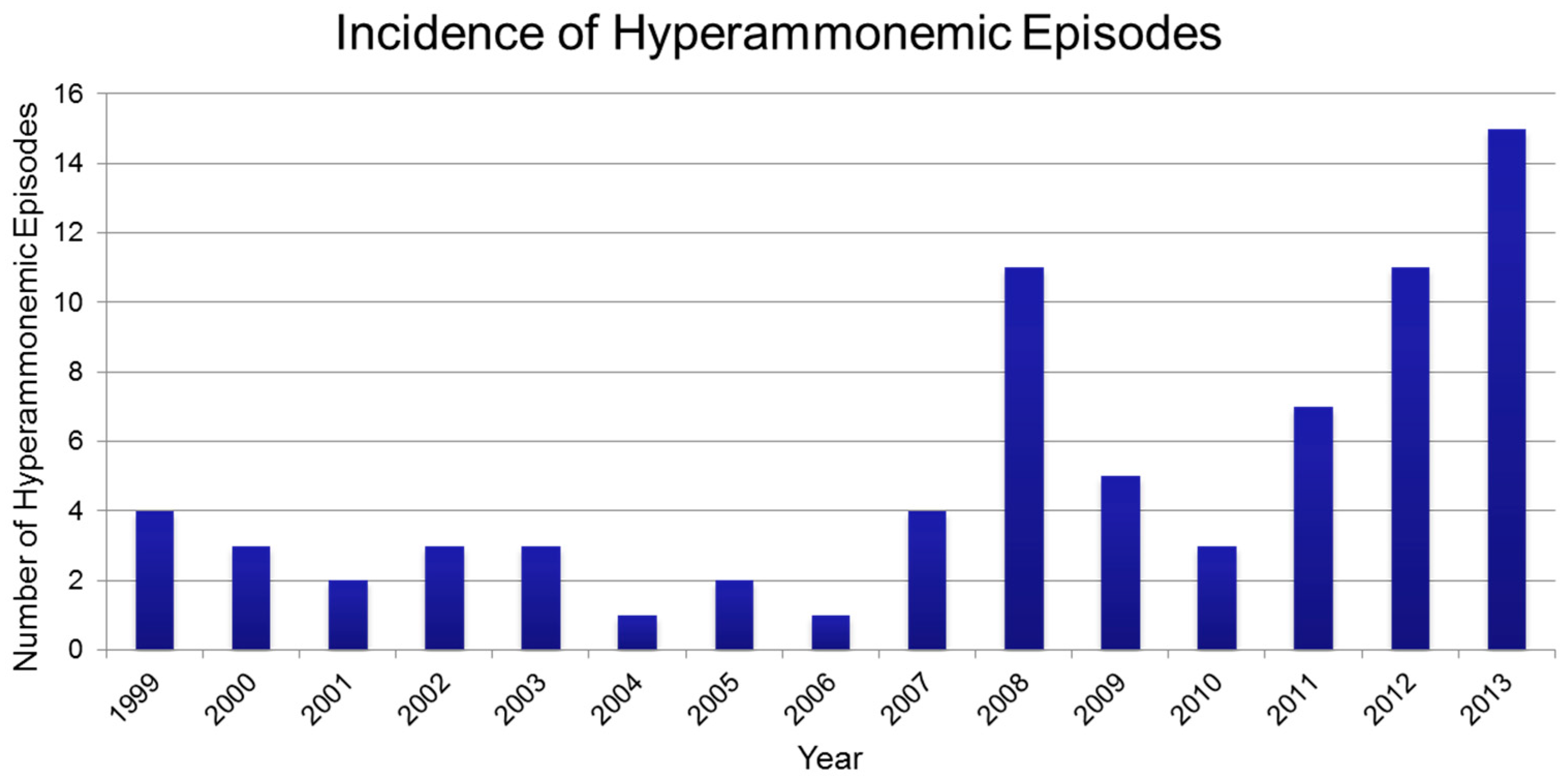
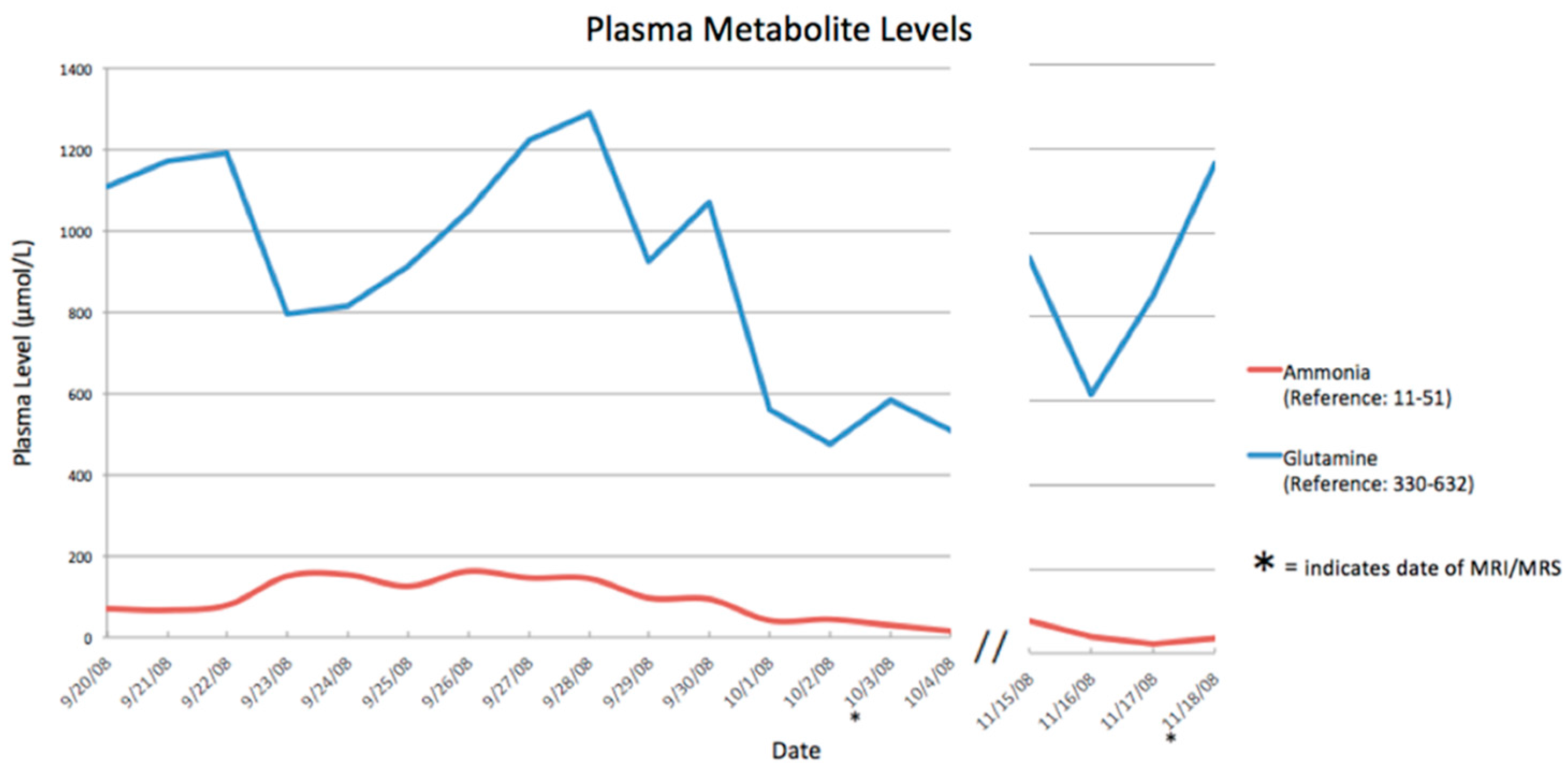
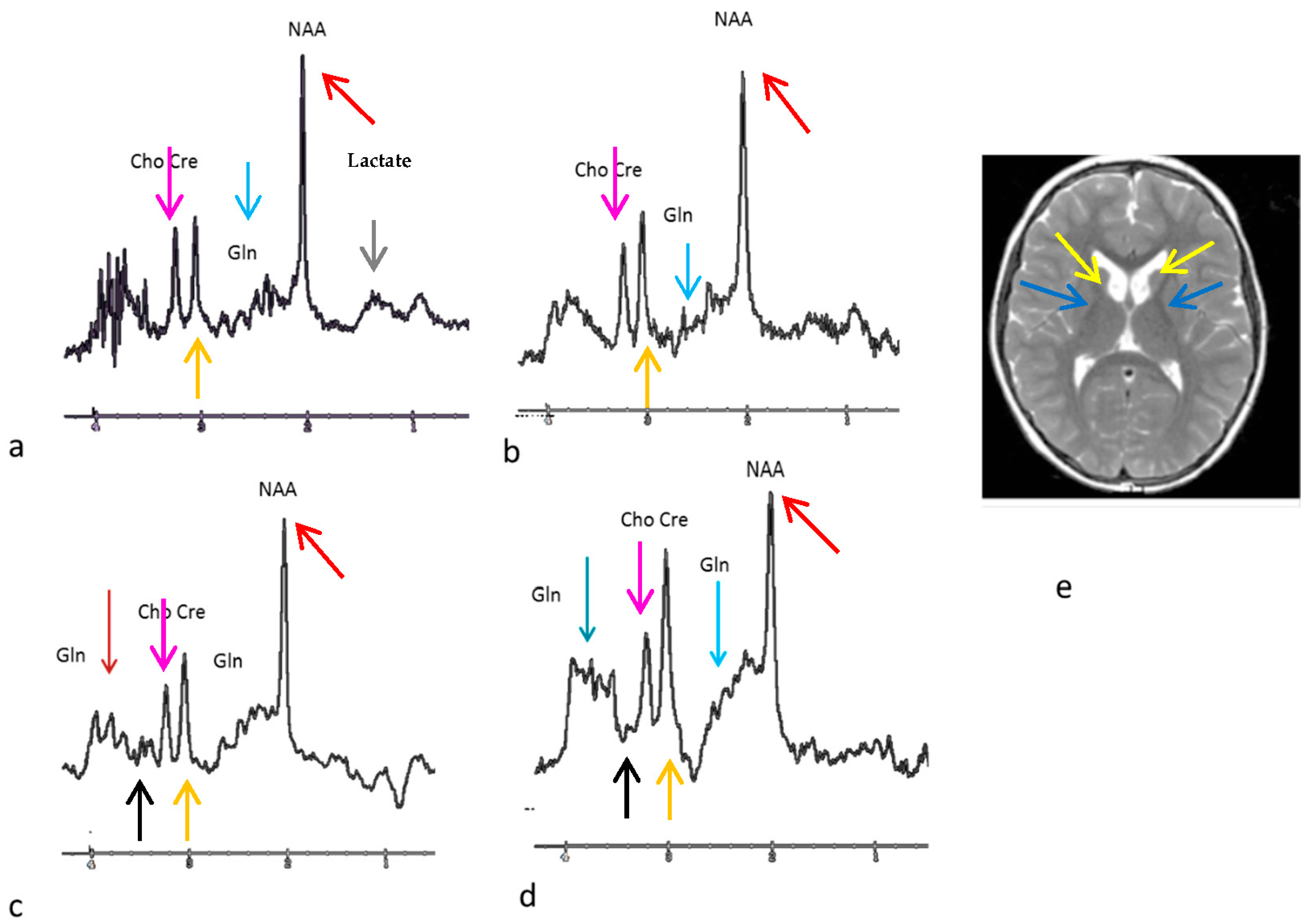
3. Results
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Applegarth, D.A.; Toone, J.R.; Lowry, R.B. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, E10. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Matsuda, I.; Oyanagi, K. Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 1991, 39, 228–229. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Rao, K.V.; Murthy, Ch.R.K.; Norenberg, M.D. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem. Int. 2006, 48, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Summar, M.; Leonard, J.V. Neurological implications of urea cycle disorders. J. Inherit. Metab. Dis. 2007, 30, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Krivitzky, L.; Babikian, T.; Lee, H.S.; Thomas, N.H.; Burk-Paull, K.L.; Batshaw, M.L. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr. Res. 2009, 66, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F.; Baumann, N.; Turpin, J.C.; Lyon-Caen, O.; Saudubray, J.M.; Cohen, D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J. Inherit. Metab. Dis. 2007, 30, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Shattuck, K.; Prust, M.J.; Seltzer, R.R.; Breeden, A.L.; Hailu, A.; Rigas, A.; Hussain, R.; VanMeter, J. Altered neural activation in ornithine transcarbamylase deficiency during executive cognition: An fMRI study. Hum. Brain Mapp. 2013, 34, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, C.; King, J.; Helman, G.; Pacheco-Colón, I.; Shattuck, K.; Breeden, A.; Seltzer, R.; VanMeter, J.W.; Gropman, A.L. Investigating neurological deficits in carriers and affected patients with ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2014, 12, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Colón, I.; Washington, S.D.; Sprouse, C.; Helman, G.; Gropman, A.L.; VanMeter, J.W. Reduced Functional Connectivity of Default Mode and Set-Maintenance Networks in Ornithine Transcarbamylase Deficiency. PLoS ONE 2015, 10, e0129595. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.I.; Pariser, A.R.; Groft, S.C.; Ishihara, R.W.; McNeil, D.E.; Tagle, D.; Griebel, D.J.; Kaler, S.G.; Mink, J.W.; Shapiro, E.G.; et al. Research challenges in central nervous system manifestations of inborn errors of metabolism. Research challenges in central nervous system manifestations of inborn errors of metabolism. Mol. Genet. Metab. 2011, 102, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.R.; Vicentini, J.; Bonilha, L.; De Campos, B.M.; Casseb, R.F.; Min, L.L. Brain Connectivity and Functional Recovery in Patients with Ischemic Stroke. J. Neuroimaging 2016. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Gertz, B.; Shattuck, K.; Kahn, I.L.; Seltzer, R.; Krivitsky, L.; VanMeter, J. Diffusion tensor imaging detects areas of abnormal white matter microstructure in patients with partial ornithine transcarbamylase deficiency. Am. J. Neuroradiol. 2010, 31, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Vafaeyan, H.; Ebrahimzadeh, S.A.; Rahimian, N.; Alavijeh, S.K.; Madadi, A.; Faeghi, F.; Harirchian, M.H.; Rad, H.S. Quantification of diagnostic biomarkers to detect multiple sclerosis lesions employing (1) H-MRSI at 3T. Australas. Phys. Eng. Sci. Med. 2015, 38, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, A.; Stricker, T.; Häberle, J. Switch from Sodium Phenylbutyrate to Glycerol Phenylbutyrate Improved Metabolic Stability in an Adolescent with Ornithine Transcarbamylase Deficiency. JIMD Rep. 2016. [Google Scholar] [CrossRef]
- Gropman, A.L.; Fricke, S.T.; Seltzer, R.R.; Hailu, A.; Adeyemo, A.; Sawyer, A.; VanMeter, J.; Gaillard, W.D.; McCarter, R.; Tuchman, M.; et al. Urea Cycle Disorders Consortium. 1H MRS identifies symptomatic and asymptomatic subjects with partial ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2008, 95, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.A.; Rao, K.V. Sparse-fur (Spf) mouse as a model of hyperammonemia: Alterations in the neurotransmitter systems. Adv. Exp. Med. Biol. 1997, 420, 143–158. [Google Scholar] [PubMed]
- Norenberg, M.D.; Rao, K.V.; Jayakumar, A.R. Mechanisms of ammonia-induced astrocyte swelling. Metab. Brain Dis. 2005, 20, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.; Colombo, J.P. Increase of tryptophan and 5-hydroxyindole acetic acid in the brain of ornithine carbamoyltransferase deficient sparse-fur mice. Pediatr. Res. 1984, 18, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Effects of hyperammonaemia on brain function. J. Inherit. Metab. Dis. 1998, 21 (Suppl. 1), 6–20. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Shiota, T.; Takei, N.; Nagashima, H. Excitatory and inhibitory amino acid neurotransmitters and ammonia metabolism in hepatic failure rats. Res. Exp. Med. 1985, 185, 399–404. [Google Scholar] [CrossRef]
- Connelly, A.; Cross, J.H.; Gadian, D.G.; Hunter, J.V.; Kirkham, F.J.; Leonard, J.V. Magnetic resonance spectroscopy shows increased brain glutamine in ornithine carbamoyl transferase deficiency. Pediatr. Res. 1993, 33, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, J.; Barkovich, A.J.; Cheng, S.F.; Kostiner, D.; Baker, J.C.; Packman, S. Brain MR imaging in acute hyperammonemic encephalopathy arising from late-onset ornithine transcarbamylase deficiency. Am. J. Neuroradiol. 2003, 24, 390–393. [Google Scholar] [PubMed]
- Gunz, C.; Choong, K.; Potter, M.; Miller, E. Magnetic resonance imaging findings and neurodevelopmental outcomes in neonates with urea cycle disorders. Int. Med. Case Rep. J. 2013, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Posset, R.; Garcia-Cazorla, A.; Valayannopoulos, V.; Teles, E.L.; Dionisi-Vici, C.; Brassier, A.; Burlina, A.B.; Burgard, P.; Cortès-Saladelafont, E.; Dobbelaere, D.; et al. Additional individual contributors of the E-IMD consortium. Age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J. Inherit. Metab. Dis. 2016, 39, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Rüegger, C.M.; Lindner, M.; Ballhausen, D.; Baumgartner, M.R.; Beblo, S.; Das, A.; Gautschi, M.; Glahn, E.M.; Grünert, S.C.; Hennermann, J.; et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J. Inherit. Metab. Dis. 2014, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Brusilow, S.W.; Cooper, A.J. Encephalopathy in acute liver failure resulting from acetaminophen intoxication: New observations with potential therapy. Crit. Care Med. 2011, 39, 2550–2553. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date (Day/Month) | Ammonia Level (Normal <50 µmol/L) |
|---|---|
| 23 September 2008 | 233 |
| 24 September 2008 | 204 |
| 25 September 2008 | 145 |
| 26 September 2008 | 194 |
| 27 September 2008 | 213 |
| 28 September 2008 | 269 |
| 29 September 2008 | 155 |
| 30 September 2008 | 145 |
| 01 October 2008 | 57 |
| 02 October 2008 | 50 |
| 27 October 2008 | 33 |
| 28 October 2008 | 22 |
| 29 October 2008 | 27 |
| 15 November 2008 | 33 |
| 16 November 2008 | 24 |
| 17 November 2008 | 22 |
| 18 November 2008 | 37 |
| Test | Age 9 Years | Age 14 Years |
|---|---|---|
| Total IQ | 73 | 74 |
| Verbal IQ | 81 | 85 |
| Performance IQ | 81 | 85 |
| Working Memory | 77 | 62 |
| Analysis Speed | 83 | 76 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourad, M.; Häberle, J.; Whitehead, M.T.; Stricker, T.; Gropman, A.L. Brain Biomarkers of Long-Term Outcome of Neonatal Onset Urea Cycle Disorder. Int. J. Neonatal Screen. 2016, 2, 10. https://doi.org/10.3390/ijns2040010
Mourad M, Häberle J, Whitehead MT, Stricker T, Gropman AL. Brain Biomarkers of Long-Term Outcome of Neonatal Onset Urea Cycle Disorder. International Journal of Neonatal Screening. 2016; 2(4):10. https://doi.org/10.3390/ijns2040010
Chicago/Turabian StyleMourad, Maha, Johannes Häberle, Matthew T. Whitehead, Tamar Stricker, and Andrea L. Gropman. 2016. "Brain Biomarkers of Long-Term Outcome of Neonatal Onset Urea Cycle Disorder" International Journal of Neonatal Screening 2, no. 4: 10. https://doi.org/10.3390/ijns2040010
APA StyleMourad, M., Häberle, J., Whitehead, M. T., Stricker, T., & Gropman, A. L. (2016). Brain Biomarkers of Long-Term Outcome of Neonatal Onset Urea Cycle Disorder. International Journal of Neonatal Screening, 2(4), 10. https://doi.org/10.3390/ijns2040010