
Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate

Abstract
: Background: Biotinidase deficiency is an inherited metabolic disorder that if untreated can result in neurological and cutaneous features. Profound biotinidase deficiency presents in early childhood with severe symptoms, whereas partial biotinidase deficiency can also present with symptoms under times of stress. Symptoms can be prevented by administering biotin. Newborn screening for the disorder is performed using dried blood spots. We examined the relationship between biotinidase activity and age at collection to determine how best to identify infants with partial biotinidase deficiency. Methods: Biotinidase activity in dried blood spots is determined using a quantitative fluorometric assay. Subsequent specimens with biotinidase activity ≤100 U were analyzed by mutation analysis to determine the range of activities expressed in infants with partial biotinidase deficiency. Results: Enzyme activity increased with age, beginning at about three days of age, and rose until plateauing at about 11 days of age. An increase of about 47.6% was observed. A total of 54 specimens had mutation analysis performed identifying 20 affected infants who would not have been identified using the original cutoff activity of 50 U. Conclusion: Biotinidase activity in infants increases with age. Age-related cutoffs assist in selectively identifying infants with partial biotinidase deficiency.1. Introduction
Biotinidase (EC 3.5.1.12) is the enzyme that cleaves biotin bound to protein to recycle free biotin from biocytin, the product the proteolytic degradation of biotinlylated proteins [1,2]. An autosomal recessively inherited disorder caused by deficient activity of biotinidase (OMIM: 253260) can produce neurological and cutaneous symptoms, including seizures, hypotonia, skin rash and alopecia, usually between the second to fifth months of life [3,4]. Many symptomatic children also have ataxia, developmental delay, conjunctivitis, hearing loss, and visual problems, including optic atrophy. Most symptomatic children develop metabolic ketoacidosis and/or organic aciduria. If untreated, some individuals develop severe metabolic compromise that can result in coma or death. Children with profound biotinidase deficiency (less than 10% of mean normal activity in serum) are effectively treated with pharmacological doses of biotin (5–20 mg daily). Early treatment can prevent the development of symptoms.
Biotinidase deficiency meets the criteria for inclusion for newborn screening [5], and is performed in all screening programs in the United States and in many other countries around the world [6]. In addition to identifying infants with profound biotinidase deficiency, infants with partial biotinidase deficiency (between 10%–30% of mean normal serum activity) have been identified. Almost all children with partial biotinidase deficiency have the D444H mutation on one allele and a mutation causing profound biotinidase deficiency on the other allele [7]. Initially, it was uncertain as to whether children with partial deficiency require biotin supplementation. Since the advent of newborn screening for the disorder, we are aware of multiple symptomatic children with partial deficiency who were not screened [8,9] or who were not treated after being identified by screening [10]. We are also aware of others who became symptomatic, but were not reported in the medical literature and improved on biotin supplementation (personal communication). Symptoms appear to occur if an individual is stressed with an infection or moderately severe gastroenteritis. Therefore, because the vitamin is safe with no known toxicity and it prevents the development of symptoms, many newborn screening programs, especially in the United States, recommend that children with partial biotinidase deficiency be treated with daily doses of 1 to 5 mg of biotin [9].
From 1987 to 2011, the Michigan Newborn Screening Laboratory screened for biotinidase activity using a modified qualitative colorimetric assay [3,11] on about 130,000 infants per year. Michigan obtains a repeat dried blood spot on infants that have a borderline biotinidase activity on the initial sample. Michigan had the highest overall detection rate of biotinidase deficiency in 2009 of seven collaborating Midwest states (Michigan, Illinois, Indiana, Kentucky, Minnesota, Ohio and Wisconsin); 1:38,540 for profound biotinidase deficiency and 1:9635 for partial biotinidase deficiency; with an overall detection rate of 1:7708.
In 2011, the Michigan Newborn Screening Laboratory switched to the semi-quantitative fluorescent PerkinElmer Neonatal Biotinidase Kit [12]. Initially, a single cutoff of 50 U was implemented for both the initial and repeat specimens. It became apparent that the enzymatic activity of most borderline positive specimens increased slightly on the repeat sample and were above the cutoff. There was concern that these infants may have partial biotinidase deficiency. In this study, we examined the range of biotinidase activities over time for individuals with partial biotinidase deficiency confirmed by genotyping to determine if we could refine the methodology of identifying infants with partial biotinidase deficiency in order to decrease the false–positive rate associated with detection of heterozygotes for profound deficiency and homozygotes for the D444H mutation.
2. Material and Methods
Biotinidase activity in newborn screening dried blood spot specimens was determined using the PerkinElmer Neonatal Biotinidase Kit [12]. It is a semi-quantitative fluorometric assay that measures the conversion of biotinyl-6-aminoquinoline to 6-aminoquinoline [13], using a Victor™ fluorometer (excitation central wavelength is 355 nm and emission central wavelength is 405 nm). Biotinidase activity is reported in enzyme units (U), where 1 U is equal to 1 nmol of end-product (1 min × dL blood).
DNA was extracted from dried blood spots using the TREC (T-Cell Receptor Excision Circle) extraction assay [14] in the newborn screening laboratory. The isolated DNA was transported on dry-ice to the DNA Diagnostic Laboratory at Henry Ford Hospital. Mutation analysis was performed using bidirectional Sanger sequencing of the biotinidase (BTD) gene exons and flanking intronic regions [14]. The 18 equivocal specimens selected for mutation analysis were collected from September 2011 through September 2012.
Approval for this method quality improvement study was obtained from the Michigan Department of Community Health Institutional Review Board, MDCH IRB LOG# 201207-07-NR.
3. Results
Determination of Biotinidase Activity Cutoffs
Strongly positive (S+, infants directly referred for diagnostic testing) and borderline positive (B+, infants requiring a repeat dried blood spot specimen) cutoff activities were established for the PerkinElmer Neonatal Biotinidase Kit by testing 5000 dried blood spots from normal infants and 28 dried blood spots from infants confirmed to have biotinidase deficiency.
These dried blood spot samples were stored at −20 °C within a week of testing. The S+ cutoff activity was established at 34 U (0.01 percentile); above the enzyme activities of all infants confirmed to have profound biotinidase deficiency. The original borderline positive cutoff was established at 50 U; above the enzyme activities of all infants confirmed to have partial biotinidase deficiency; this cutoff was later changed to 65 U per laboratory quality assurance protocol. An individual was considered equivocal and re-assayed if below 90 U.
Infants with a B+ initial sample and B+ repeat sample are referred for confirmatory genotyping. Frequently, the activity in repeat specimens was higher than the activity of the initial specimens and was reported as normal. Analyses of enzyme activities of repeat samples indicated that biotinidase activity increased with the age of the infant. The relationship of biotinidase activity and age is shown in Figure 1. This graph represents the mean biotinidase enzyme activity at a given age of collection in days (n > 60,000 samples from 2011–2012). Enzyme activity increased with age, beginning at approximately three days of age, and steadily rose to a plateau at approximately 11 days of age.
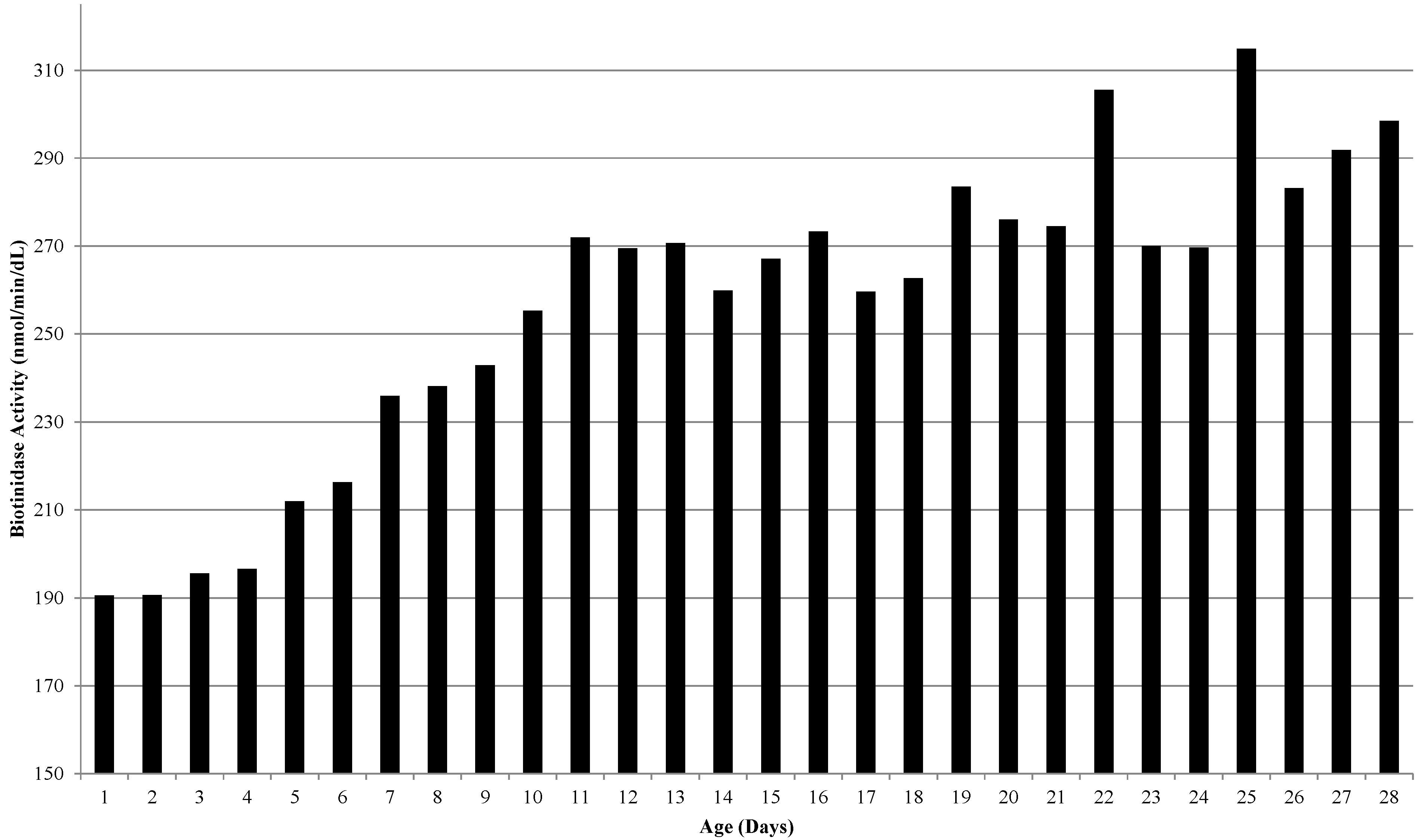
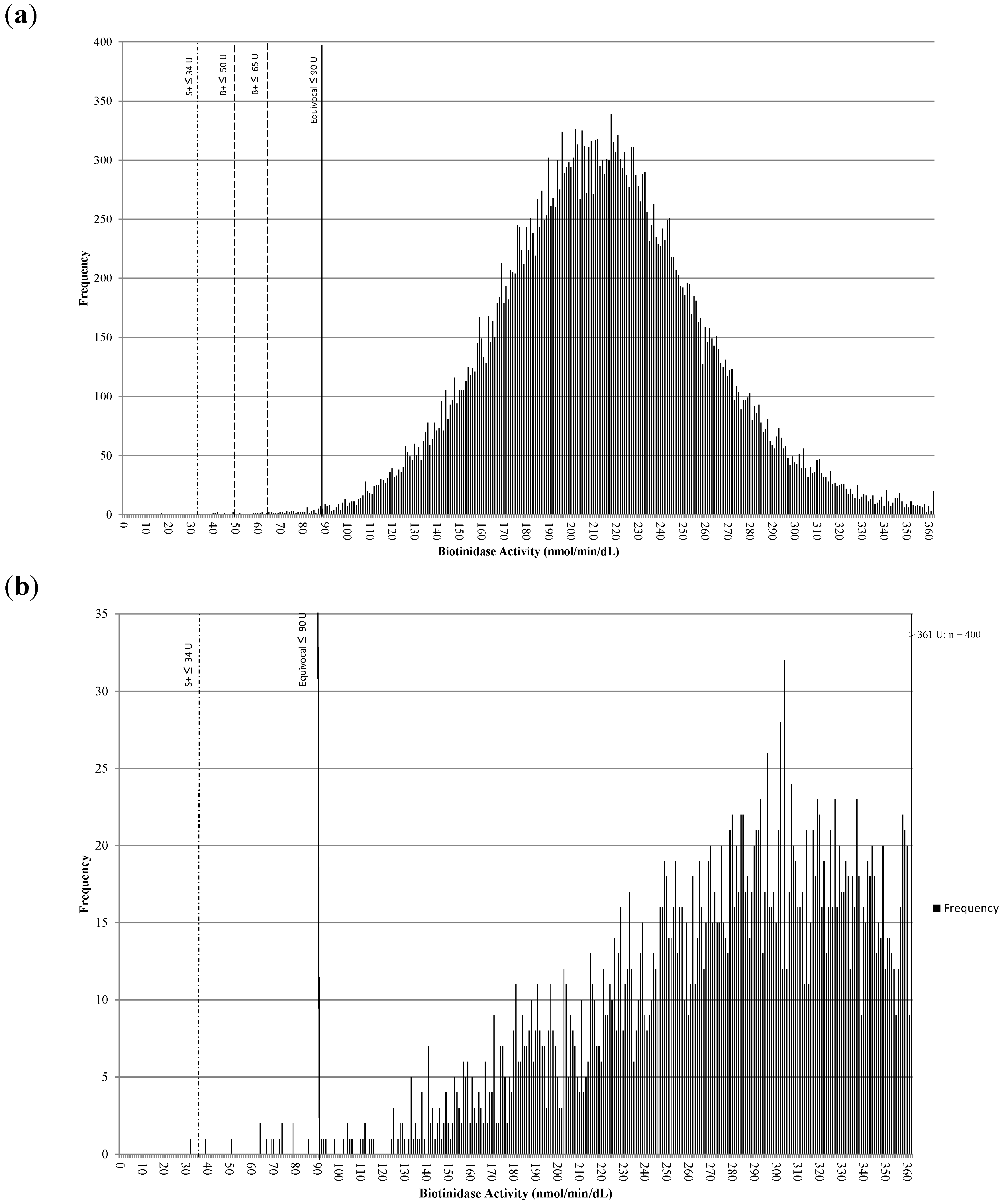
Figure 2a is a histogram showing the distribution of biotinidase activities for samples collected at ≤72 h of age (n > 30,000 from 2013). The S+ cutoff was 34 U, the original B+ cutoff was 50 U, the revised B+ cutoff was 65 U, and the equivocal cutoff was 90 U. Figure 2b is a histogram showing the distribution of biotinidase activities for samples collected at >72 h of age (n > 3000 from 2013). The S+ cutoff was 34 U and the equivocal cutoff was 90 U. The mean activity increased from 210 U at collection of ≤72 h old to 310 U at collection of >72 h old; a net increase of about 100 U or 47.6%.
Infants with activities below the cutoffs are referred for diagnostic evaluations which usually required genotyping. Infants with activities ≤65 U on the repeat sample were referred for diagnostic testing in accordance with laboratory protocol. To establish a clinically valid cutoff for samples >72 h old, it was necessary to identify all infants with partial biotinidase deficiency. The preliminary age-appropriate biotinidase activity cutoff, using 30% of the mean for samples collected at greater than 72 h of age, was 93 U, which was rounded to 100 U. All repeat samples not referred for diagnostic testing with biotinidase activity below this cutoff had mutation analysis of the BTD gene performed. It was hypothesized that these samples would include those with partial biotinidase deficient genotypes, homozygous for D444H and heterozygous for a single mutation for profound biotinidase deficiency. Eighteen repeat samples with activities between 66 U and 100 U had mutation analysis performed. Of these samples, five had confirmed partial biotinidase deficiency (two with known mutations causing profound deficiency on one allele and the D444H mutation on the other allele and two with novel mutations, R209H (c.626G>A) and T196A (c.586A>G) shown to cause profound deficiency on one allele and the D444H mutation on the other allele) [10,15]. These results suggest that heterozygotes with a single mutation for profound deficiency have activities >100 U on repeat specimens. In addition to these five individuals with partial biotinidase deficiency, 13 were homozygous for the D444H mutation. None of the samples sent for mutation analysis (≤100 U) were heterozygous for a single mutation for profound biotinidase deficiency.
Partial biotinidase activity was identified in 20 infants who had repeat biotinidase activity above the initially identified cutoff of 50 U. This analysis confirmed that infants with partial biotinidase deficiency, while abnormal on the initial screen, were above the initial cutoff on a repeat sample.
Analysis of the increase in biotinidase activity with age also indicates that the increase is genotype dependent. The increase in enzyme activity for infants with partial biotinidase deficiency was, in general, less than that for infants homozygous for the D444H mutation. An age-based cutoff was implemented to optimize the identification of infants with partial biotinidase deficiency. Combining a borderline positive repeat sample protocol together with an age-related enzyme activity cutoff was an effective method of selectively identifying infants with partial biotinidase deficiency.
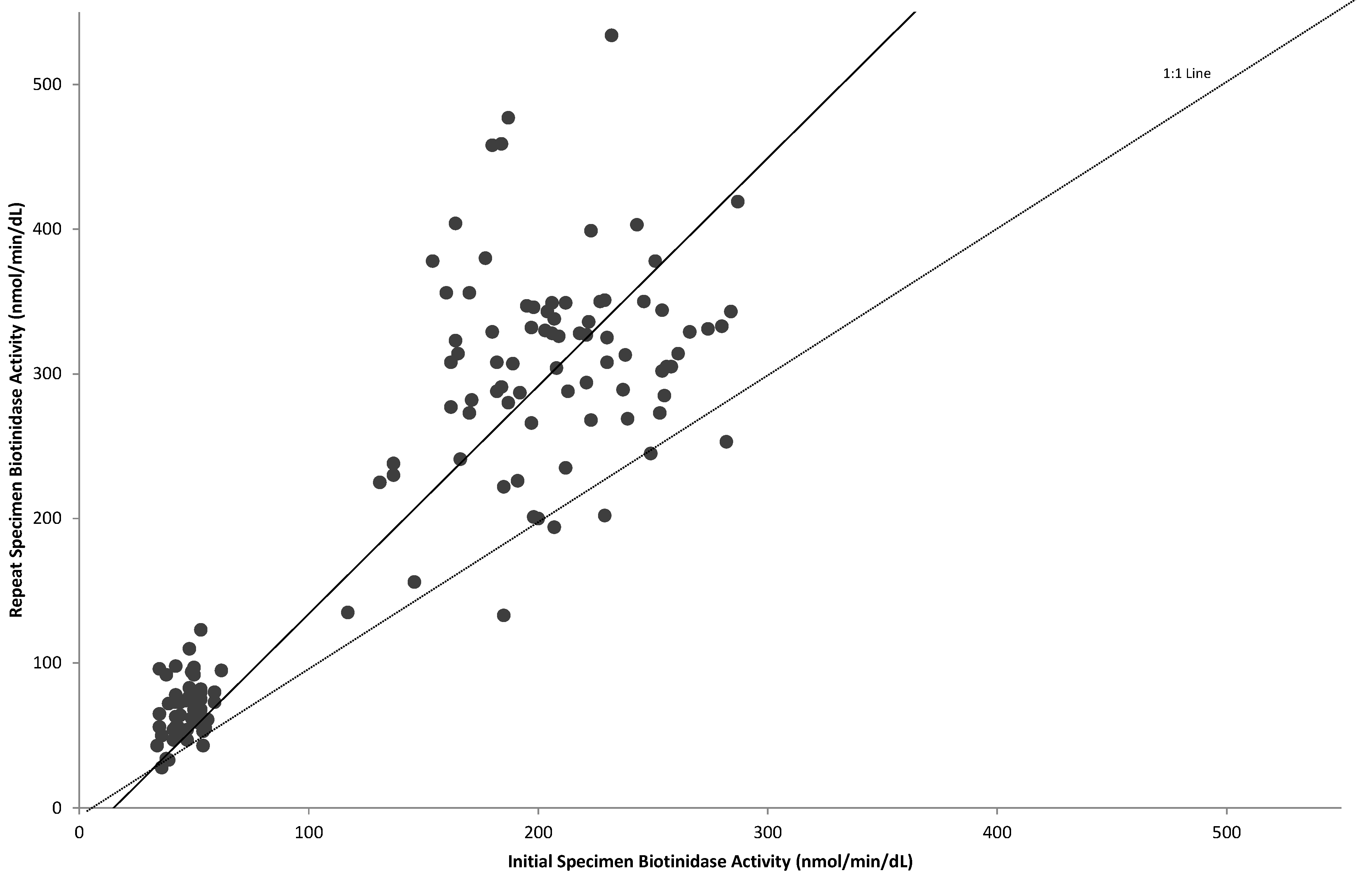
The activities of initial specimens are compared to those of repeat specimens in Figure 3. In general, biotinidase activity increased by a factor of 1.6 on the repeat specimen. A similar scatter-graph including only infants confirmed as having partial deficiency, one with profound biotinidase deficiency and those homozygous for the D444H mutation are shown in Figure 4. From this graph, it is evident that the enzyme activities of infants homozygous for the D444H mutation overlap significantly on the initial screen with infants with partial biotinidase deficiency. It was assumed that infants homozygous for the D444H mutation have activities similar to those heterozygous with a single mutation for profound biotinidase deficiency; however, the study did not identify any individuals heterozygous for a single mutation for profound biotinidase deficiency. Due to the overlap of activities on the initial screen, it is not possible to eliminate infants homozygous for the D444H mutation and still identify infants with partial biotinidase deficiency using the initial samples. Most infants confirmed to have partial biotinidase deficiency had repeat sample activities below 80 U; therefore, the age-related repeat cutoff was set at 80 U. With this cutoff we are able minimize the number of infants homozygous for the D444H mutation who are referred for further diagnostic evaluation and genotyping.
Overlap exists between infants with profound deficiency, partial deficiency and those homozygous for the D444H mutation. Infants with profound biotinidase deficiency can have initial activities in the B+ range. Enzyme activities for all infants confirmed to have profound biotinidase deficiency, partial biotinidase deficiency and those homozygous for the D444H mutation are shown in Table 1 (data from August 2011 to July 2013). Notably there is overlap in activities of infants with partial biotinidase deficiency with those homozygous for the D444H mutation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case # | Biotinidase Activity, U | Allele 1 | Allele 2 | Genotypic Classification | |
|---|---|---|---|---|---|
| Initial | Repeat | ||||
| 1 | 30 | _ | P497S/Y428Y | P497S/Y428Y | Profound Biotinidase Deficiency |
| 2 | 23 | _ | A162V | Q456H | |
| 3 | 25 | _ | Unknown* | Unknown* | |
| 4 | 36 | 50 | Unknown* | Unknown* | |
| 5 | 34 | _ | P235S | D444H | Partial Biotinidase Deficiency |
| 6 | 31 | _ | I108V | D444H | |
| 7 | 30 | _ | Q456H | D444H | |
| 8 | 29 | _ | A171T/D444H | D444H | |
| 9 | 30 | _ | L215F | D444H | |
| 10 | 39 | 72 | Q456H | D444H | |
| 11 | 42 | 63 | Q456H | D444H | |
| 12 | 50 | 79 | c.933delT | D444H | |
| 13 | 44 | 73 | R211C | D444H | |
| 14 | 44 | 64 | Q456H | D444H | |
| 15 | 43 | 56 | Unknown* | Unknown* | |
| 16 | 55 | 61 | Unknown* | Unknown* | |
| 17 | 53 | 68 | R157H | D444H | |
| 18 | 35 | 56 | Y414-V417del | D444H | |
| 19 | 54 | 43 | Unknown* | Unknown* | |
| 20 | 48 | 77 | R209H | D444H | |
| 21 | 38 | 34 | A171T/D444H | D444H | |
| 22 | 34 | 43 | A171T/D444H | D444H | |
| 23 | 49 | 62 | R211C | D444H | |
| 24 | 52 | 59 | R538C | D444H | |
| 25 | 56 | 61 | Q456H | D444H | |
| 26 | 51 | 79 | G45R/A100T | D444H | |
| 27 | 42 | 56 | A171T/D444H | D444H | |
| 28 | 54 | 53 | Q456H | D444H | |
| 29 | 36 | 28 | A171T/D444H | D444H | |
| 30 | 53 | 82 | Y454C | D444H | |
| 31 | 41 | 47 | A171T/D444H | D444H | |
| 32 | 50 | 68 | Unknown* | Unknown* | |
| 33 | 35 | 65 | A171T/D444H | D444H | |
| 34 | 41 | 54 | Y414-V417del | D444H | |
| 35 | 39 | 33 | A171T/D444H | D444H | |
| 36 | 47 | 47 | Q456H | D444H | |
| 37 | 42 | 50 | Q456H | D444H | |
| 38 | 49 | 94 | T196A | D444H | |
| 39 | 32 | _ | D444H | D444H | Homozygous for D444H Mutation |
| 40 | 42 | 78 | D444H | D444H | |
| 41 | 62 | 95 | D444H | D444H | |
| 42 | 42 | 98 | D444H | D444H | |
| 43 | 50 | 97 | D444H | D444H | |
| 44 | 54 | 60 | D444H | D444H | |
| 45 | 53 | 80 | D444H | D444H | |
| 46 | 42 | 73 | D444H | D444H | |
| 47 | 35 | 96 | D444H | D444H | |
| 48 | 54 | 59 | D444H | D444H | |
| 49 | 42 | 76 | D444H | D444H | |
| 50 | 53 | 75 | D444H | D444H | |
| 51 | 50 | 92 | D444H | D444H | |
| 52 | 38 | 92 | D444H | D444H | |
| 53 | 46 | 74 | D444H | D444H | |
| 54 | 59 | 73 | D444H | D444H | |
Highlighted Samples have activities greater than the revised repeat activity cutoff of 65 U. *Unknown: Genotypes were not reported to the Newborn Screening Laboratory.

Infants with profound deficiency had activities between 23 and 36 U on the initial specimen and up to 50 U on a repeat specimen. Infants with partial deficiency had activities between 29 and 56 U on the initial specimen and up to 82 U on the repeat specimen. Infants who are homozygous for the D444H mutation had activities between 32 and 62 U on initial specimens and up to 98 U on repeat specimens. It is probable that some infants homozygous for the D444H mutation will have activities greater than 100 U on repeat specimens, although 98 U was the highest repeat activity of specimens from infants confirmed to be homozygous for the D444H mutation in this study. Because overlap is evident between the enzyme-deficient infants, it is likely that overlap also exists throughout the genotypic spectrum. This overlap makes it difficult, if not impossible, to accurately classify genotypes by activity. Consequently, some infants homozygous for the D444H mutation will be referred for diagnostic testing, along with those with partial biotinidase deficiency. However, this method should decrease the number of infants who are homozygous for the D444H mutation and would have been referred for diagnostic evaluation while actively screening for partial biotinidase deficiency.
Based on the results of this study, we have set our borderline positive cutoff at 80 U for specimens from infants greater than 72 h of age. This allows us to identify the majority of infants with partial deficiency. This age-related cutoff activity, in conjunction with the revised initial B+ cutoff, would have allowed us to identify 20 additional infants with partial biotinidase deficiency that would not have been identified using the original cutoff activity of 50 U (Table 1). This cutoff does not eliminate, but should decrease, the number of false-positive infants who are homozygous for the D444H mutation and are be referred for genotyping (Figure 4). Ten of the confirmed infants homozygous for the D444H mutation would have been referred for diagnostic testing using the established age-related cutoff.
4. Discussion
The Michigan Newborn Screening Laboratory used the PerkinElmer fluorometric biotinidase activity assay to acquire semi-quantitative data to facilitate the classification of infants with biotinidase deficiency. This allowed us to set cutoff activities to identify profound and partial biotinidase deficiency in our screening population and to improve our performance metrics. Our detection rate improved from 1:14,958 in 2008 to 1:6197 in 2012. This study shows that biotinidase activity increases with age during infancy. According to Michigan’s infant population data, the increase begins at about the third day of life. Because of this increase in activity, we were concerned about the sensitivity of a single borderline positive cutoff activity for identifying partial biotinidase deficiency. It appeared likely that infants with partial biotinidase deficiency had activities that were above the initially determined cutoff activity on a repeat specimen.
Using the data gathered from mutation analyses and other confirmed cases, the age-related cutoff activity of 80 U is more effective at identifying infants with partial biotinidase deficiency when the samples are collected at greater than 72 h of age. The preliminary calculated cutoff activity of 93 U was not used because of the overlap between infants with partial biotinidase deficiency and those homozygous for the D444H mutation. Employing a cutoff of 80 U for samples collected from infants greater than 72 h of age is a compromise between maximizing partial biotinidase deficiency identification and minimizing detection of infants homozygous for the D444H mutation. These data suggest that most infants with repeat results greater than, but near 80 U, are likely homozygous for the D444H mutation.
This study demonstrates that age-related cutoffs can greatly improve the ability to identify infants with partial biotinidase deficiency and decrease the false-positive rate of screening for this disorder. In the future, with decreasing cost and increasing speed of mutation analysis, it may be possible to decrease the false-positive rate further by incorporating a second tier mutation analysis.
Acknowledgments
The authors would like to thank Heather Wood, Kevin Cavanagh, Mary Kleyn, and Karen Andruszewski. This work was supported in part by the Safra Research Fund (B.W.).
Authors Contributions
Nicole VanVleck and Mary Seeterlin conceived of the idea for the research and analyzed the data. Nicole also wrote the manuscript. Barry Wolf was involved with the analysis, clinical interpretation of the data and writing of the manuscript. Kristin Monaghan performed the mutation analyses. Eleanor Stanley, Harry Hawkins and Bonita Taffe are the supervisors of the laboratory and assisted in the writing of the manuscript.
Conflicts of Interest
All of the authors declare no conflict of interest.
References
- Thoma, R.W.; Peterson, W.H. The enzymatic degradation of soluble bound biotin. J. Biol. Chem. 1954, 210, 569–579. [Google Scholar] [PubMed]
- Pispa, J. Animal biotinidase. Ann. Med. Exp. Biol. Fenn. 1965, 43 (Suppl. 5), 1–39. [Google Scholar] [PubMed]
- Wolf, B.; Grier, R.E.; Allen, R.J.; Goodman, S.I.; Kien, C.L. Biotinidase deficiency: The enzymatic defect in late-onset multiple carboxylase deficiency. Clin. Chim. Acta 1983, 131, 273–281. [Google Scholar] [CrossRef]
- Wolf, B.; Heard, G.S.; Weissbecker, K.A.; Secor McVoy, J.R.; Grier, R.E.; Leshner, R.T. Biotinidase deficiency: Initial clinical features and rapid diagnosis. Ann. Neurol. 1985, 18, 614–617. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B. Worldwide survey of neonatal screening for biotinidase deficiency. J. Inherit. Metab. Dis. 1991, 14, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B. The neurology of biotinidase deficiency. Mol. Genet. Metab. 2011, 104, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Swango, K.L.; Demirkol, M.; Huner, G.; Pronicka, E.; Sykut-Cegielska, J.; Schulze, A.; Wolf, B. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum. Genet. 1998, 102, 571–575. [Google Scholar] [CrossRef] [PubMed]
- McVoy, J.R.; Levy, H.L.; Lawler, M.; Schmidt, M.; Ebers, D.D.; Hart, P.S.; Pettit, D.; Blitzer, M.G.; Wolf, B. Partial biotinidase deficiency: Clinical and biochemical features. J. Pediatr. 1990, 116, 78–83. [Google Scholar] [CrossRef]
- Wolf, B. Why perform newborn screening for profound and partial biotinidase deficiency? Mol. Genet. Metab. 2015. [Google Scholar] [CrossRef]
- Jay, A.M.; Conway, R.L.; Feldman, G.L.; Nahhas, F.; Spencer, L.; Wolf, B. Outcomes of individuals with profound and partial biotinidase deficiency ascertained by newboorn screening in Michigan over 25 years. Genet. Med. 2014. [Google Scholar] [CrossRef]
- Heard, G.S.; Secor McVoy, J.R.; Wolf, B. A screening method for biotinidase deficiency in newborns. Clin. Chem. 1984, 30, 125–127. [Google Scholar] [PubMed]
- PerkinElmer Neonatal Biotinidase Kit Insert; Wallac Oy: Mustionkatu, Turku, Finland, 2010.
- Marklund, N.; Morales, D.; Clausen, F.; Hanell, A.; Kiwanuka, O.; Pitkanen, A.; Gimbel, D.A.; Philipson, O.; Lanfelt, I.; Hellered, L.; et al. Functional outcome is impaired following traumatic brain injury in aging Nogo-A/B- deficient mice. Neuroscience 2009, 163, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Norrgard, K.J.; Pomponio, R.J.; Hymes, J.; Wolf, B. Mutations causing profound biotinidase deficiency in children ascertained by newborn screening in the United States occur at different frequencies than in symptomatic children. Pediatr. Res. 1999, 46, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Spencer, L.; Nahhas, F.; Miller, J.; Fribley, A.; Feldman, G.; Conway, R.; Wolf, B. Novel mutations causing biotinidase deficiency in individuals identified by newborn screening in Michigan including a unique intronic mutation that alters mRNA expression of the biotinidase gene. Mol. Genet. Metab. 2014, 112, 242–246. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
VanVleck, N.; Wolf, B.; Seeterlin, M.; Monaghan, K.G.; Stanley, E.; Hawkins, H.; Taffe, B. Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate. Int. J. Neonatal Screen. 2015, 1, 45-56. https://doi.org/10.3390/ijns1010045
VanVleck N, Wolf B, Seeterlin M, Monaghan KG, Stanley E, Hawkins H, Taffe B. Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate. International Journal of Neonatal Screening. 2015; 1(1):45-56. https://doi.org/10.3390/ijns1010045
Chicago/Turabian StyleVanVleck, Nicole, Barry Wolf, Mary Seeterlin, Kristin G. Monaghan, Eleanor Stanley, Harry Hawkins, and Bonita Taffe. 2015. "Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate" International Journal of Neonatal Screening 1, no. 1: 45-56. https://doi.org/10.3390/ijns1010045
APA StyleVanVleck, N., Wolf, B., Seeterlin, M., Monaghan, K. G., Stanley, E., Hawkins, H., & Taffe, B. (2015). Improved Identification of Partial Biotinidase Deficiency by Newborn Screening Using Age-Related Enzyme Activity Cutoffs: Reduction of the False-Positive Rate. International Journal of Neonatal Screening, 1(1), 45-56. https://doi.org/10.3390/ijns1010045