

Mutant Isocitrate Dehydrogenase 1 Expression Enhances Response of Gliomas to the Histone Deacetylase Inhibitor Belinostat

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Immunoblot Analyses
2.3. Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS)
2.4. Cell Growth and Apoptosis Assays
2.5. Clinical Trial Information
3. Results
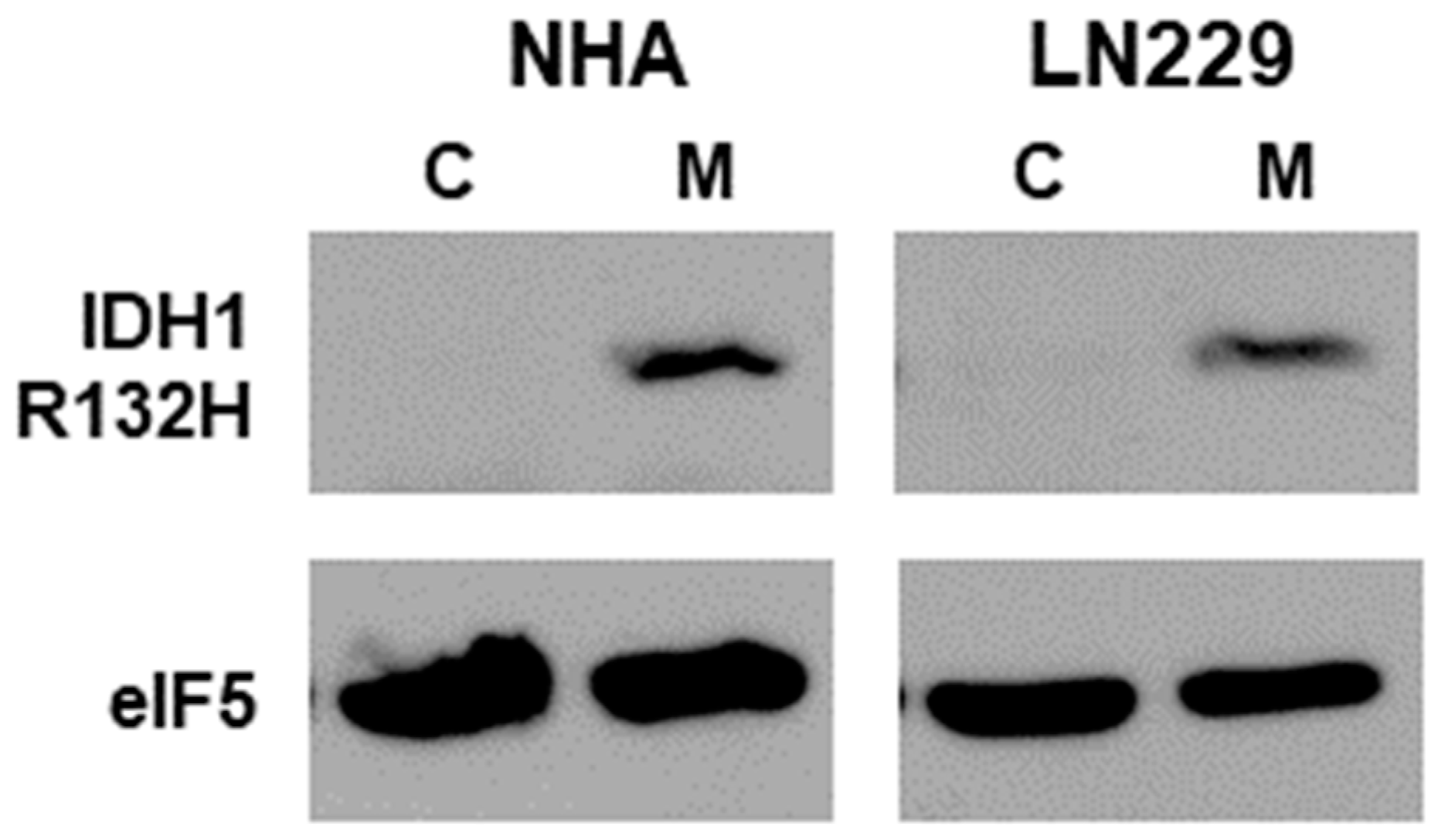
3.1. Expression of IDH1-R132H Mutant in Glioma Cell Lines
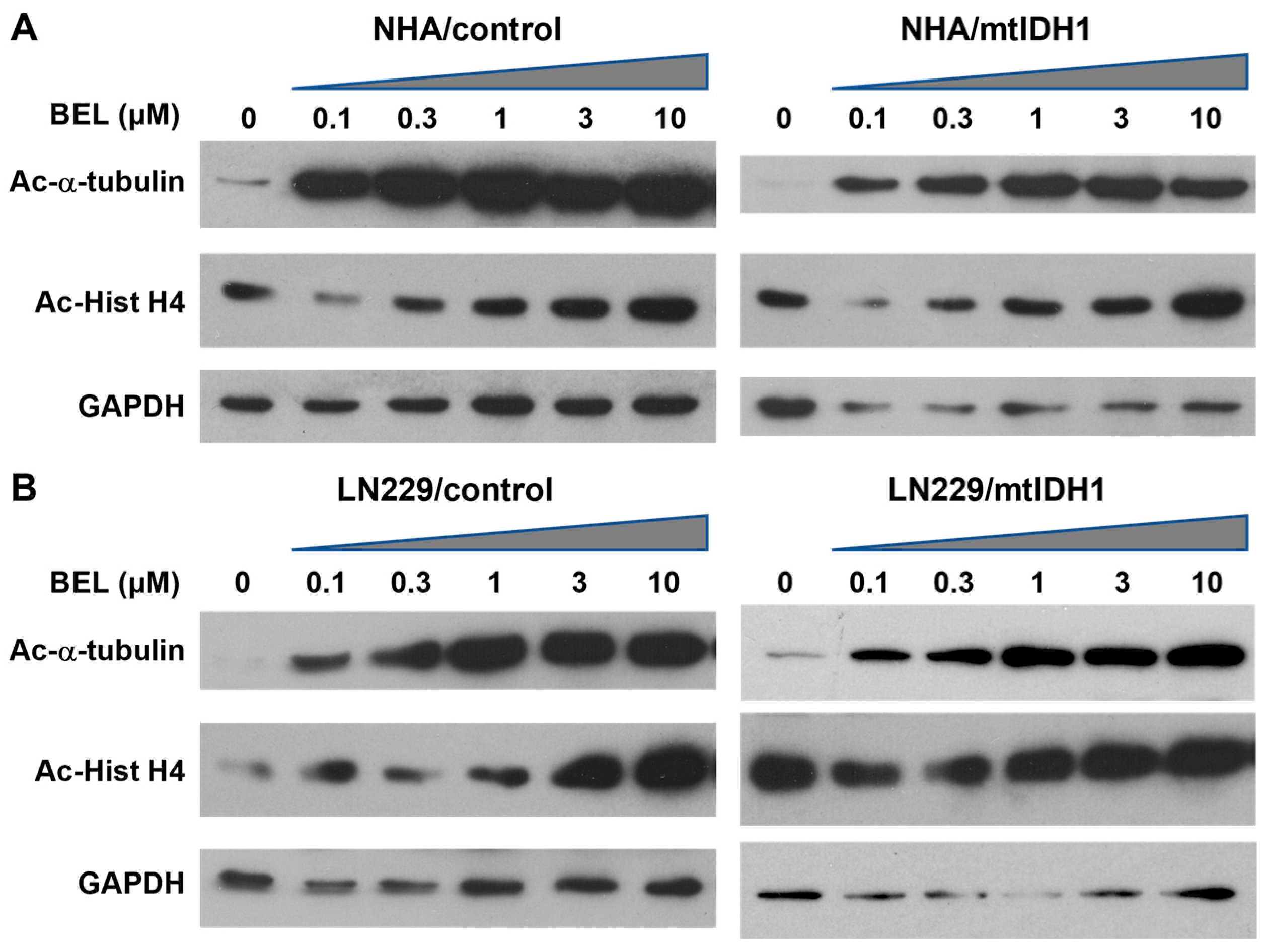
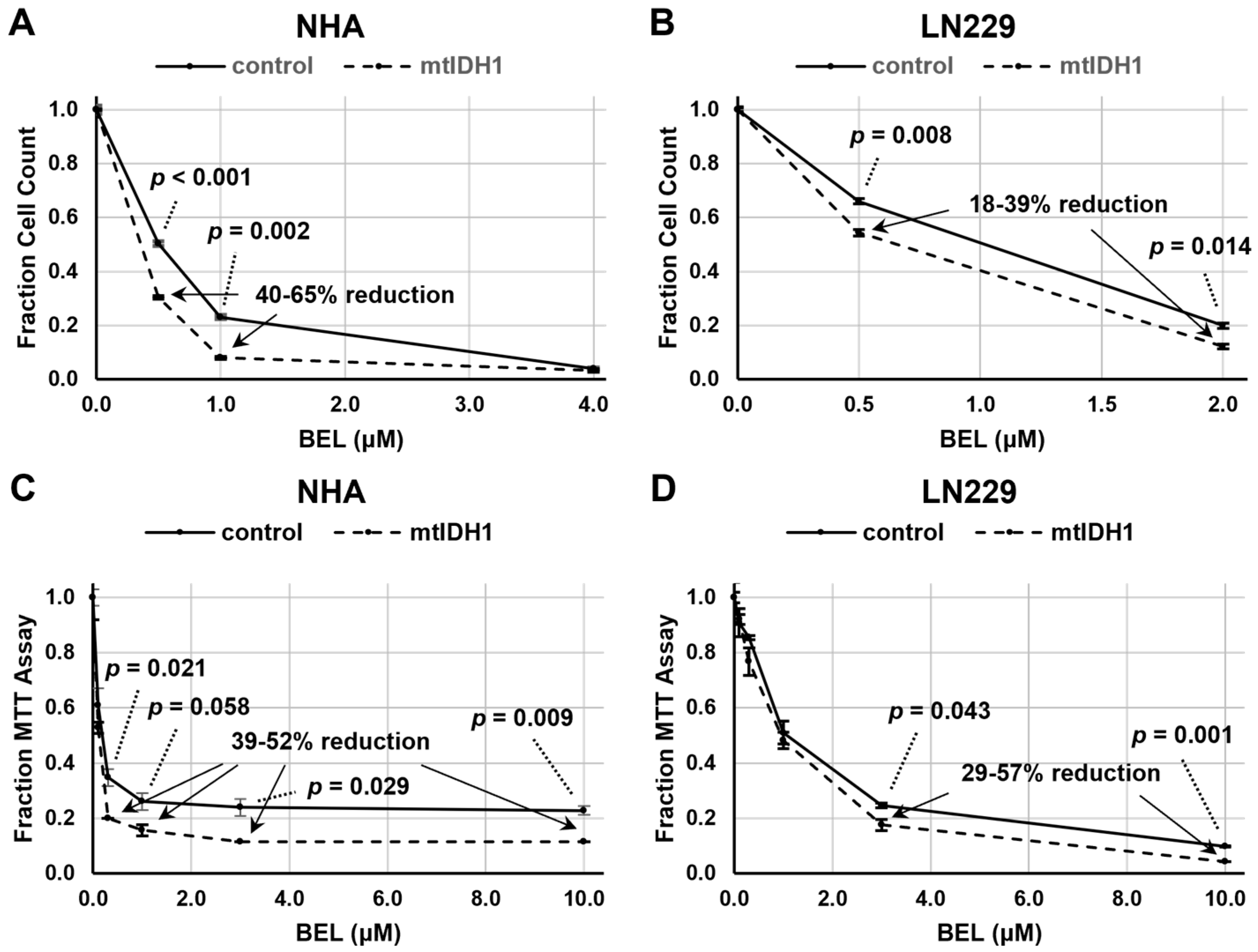
3.2. Sensitivity of mtIDH1-Expressing and Control Glioma Cells to HDAC Inhibition
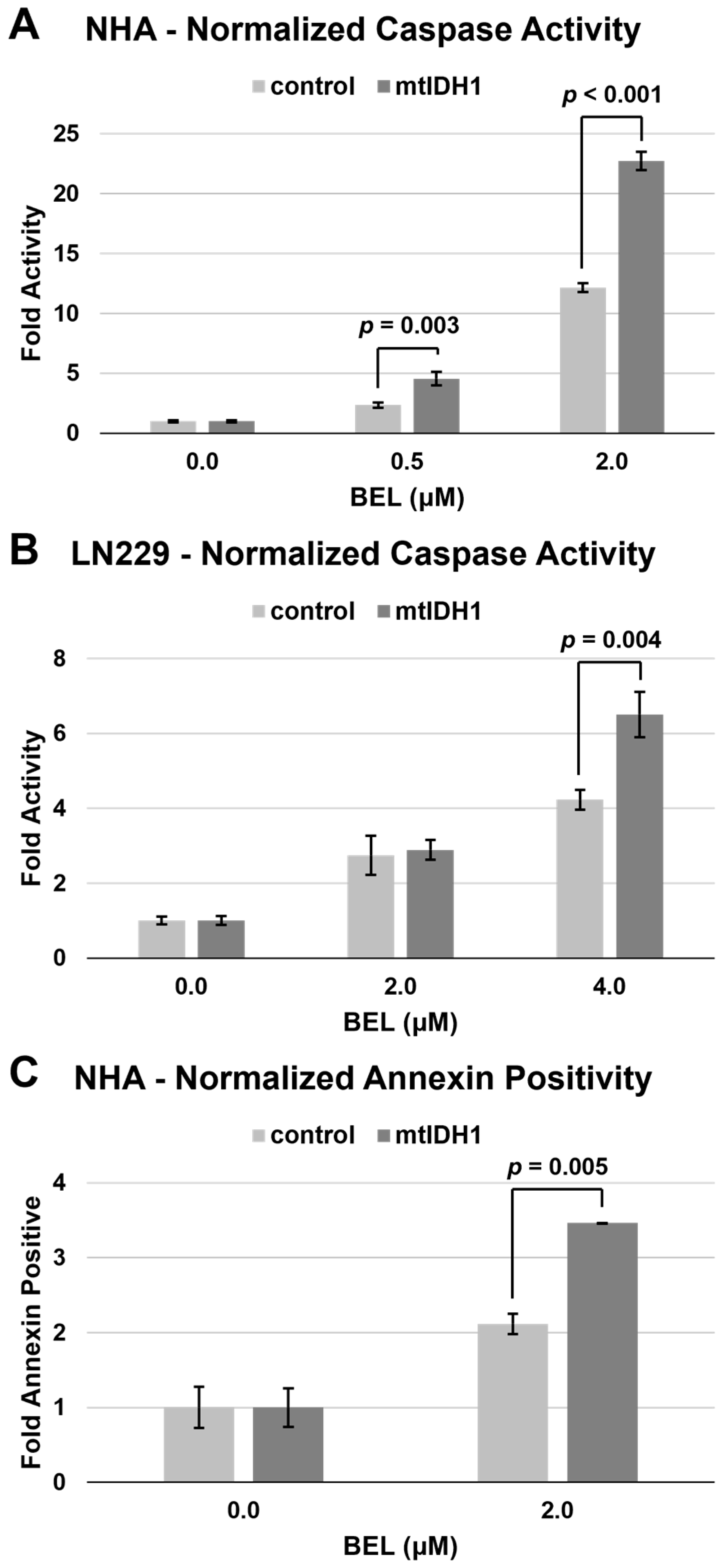
3.3. Glioma Cells Expressing mtIDH1 Display Increases Apoptosis with HDAC Inhibition
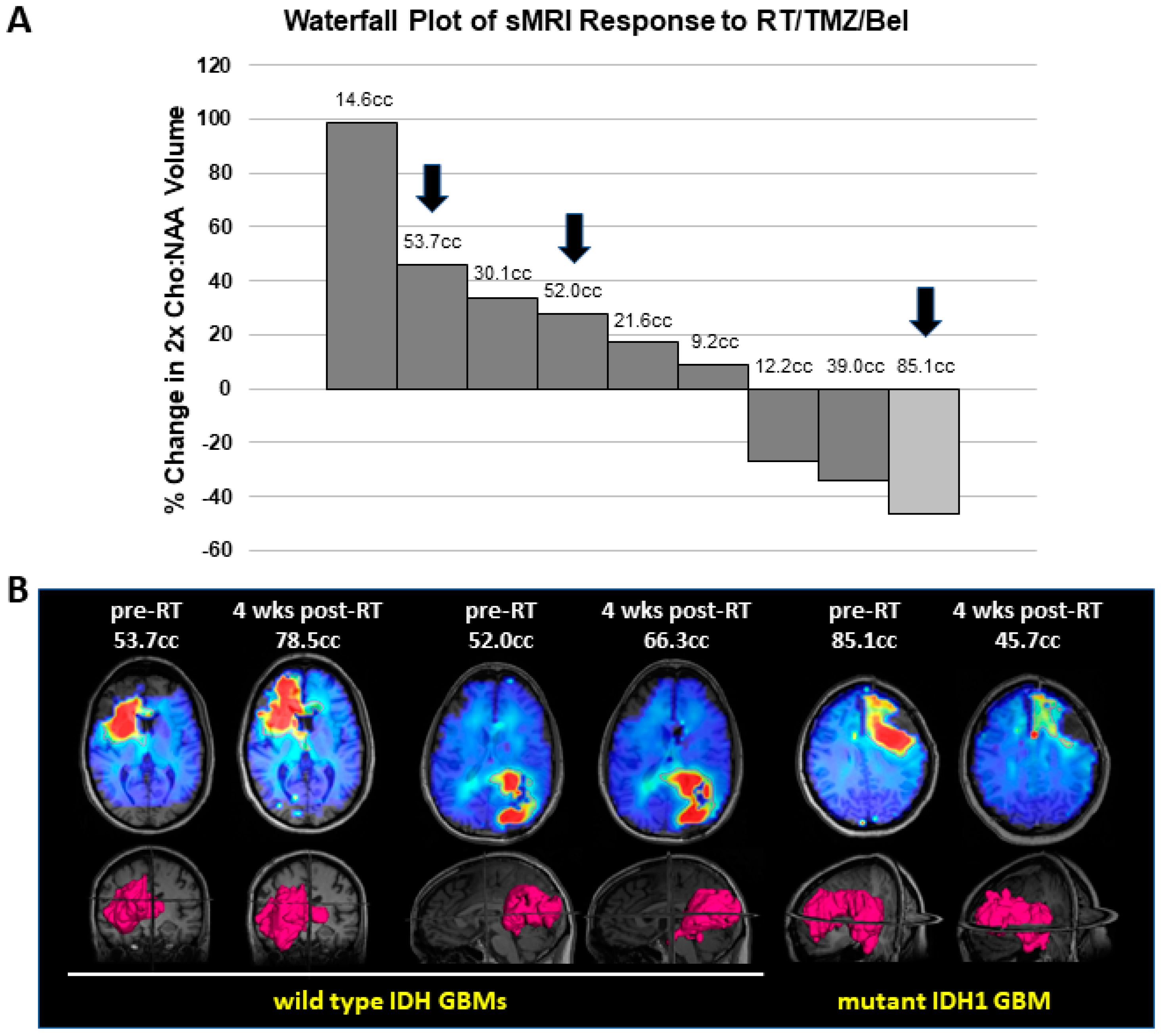
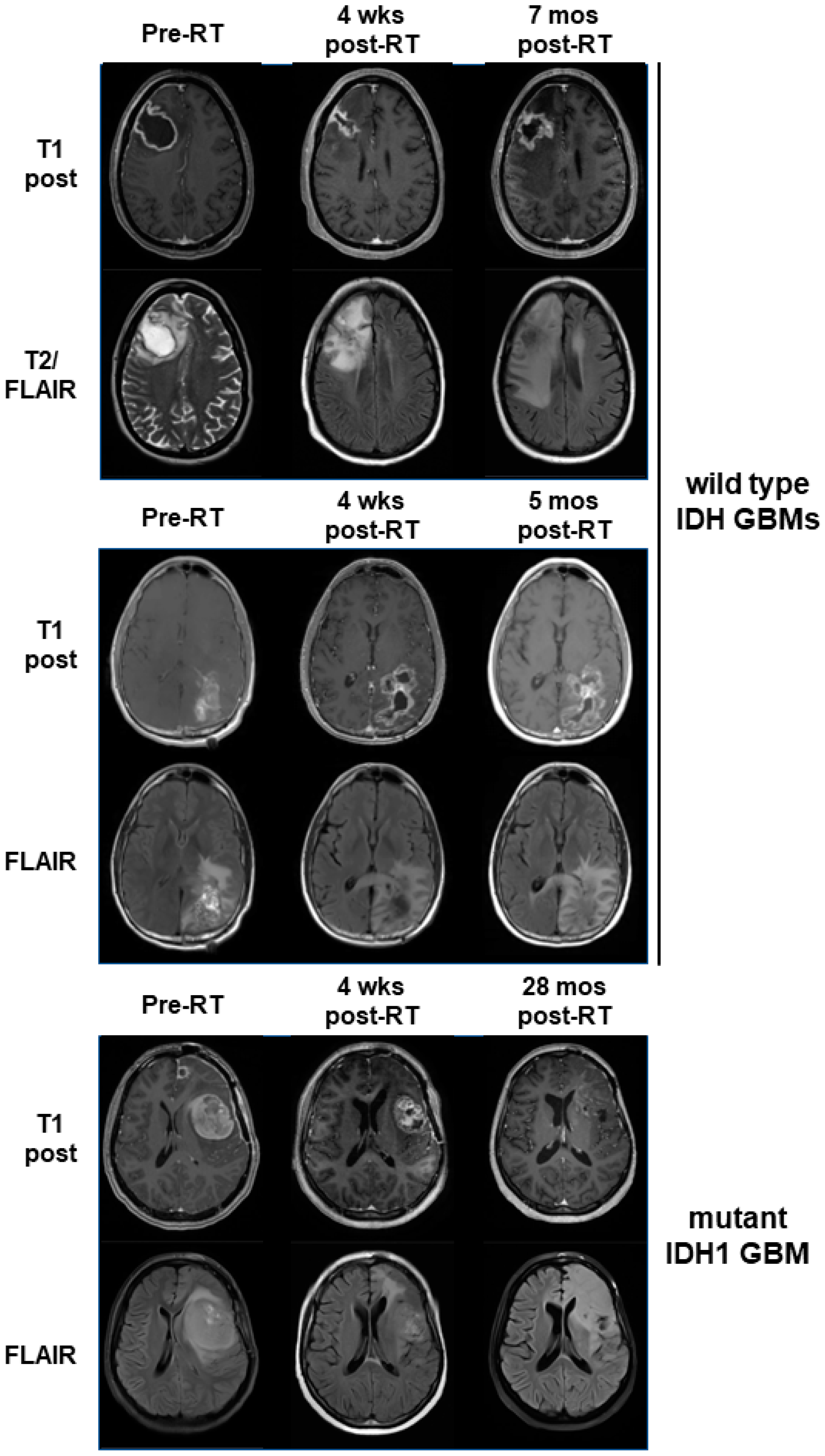
3.4. Mutant IDH1 GBM Shows Greater Response to a Belinostat-Containing Regimen than Corresponding Wild-Type IDH GBMs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Kim, G.S.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Vander Heiden, M.G.; Sorensen, A.G. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci. Transl. Med. 2012, 4, 116ra14. [Google Scholar] [CrossRef] [PubMed]
- Elkhaled, A.; Jalbert, L.E.; Phillips, J.J.; Yoshihara, H.A.I.; Parvataneni, R.; Srinivasan, R.; Bourne, G.; Berger, M.S.; Chang, S.M.; Cha, S.; et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci. Transl. Med. 2012, 4, 116ra115. [Google Scholar] [CrossRef]
- Kalinina, J.; Carroll, A.; Wang, L.; Yu, Q.; Mancheno, D.E.; Wu, S.; Liu, F.; Ahn, J.; He, M.; Mao, H.; et al. Detection of “oncometabolite” 2-hydroxyglutarate by magnetic resonance analysis as a biomarker of IDH1/2 mutations in glioma. J. Mol. Med. 2012, 90, 1161–1171. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Branzoli, F.; Marjanska, M. Magnetic resonance spectroscopy of isocitrate dehydrogenase mutated gliomas: Current knowledge on the neurochemical profile. Curr. Opin. Neurol. 2020, 33, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, P.; Chaumeil, M.M.; Ronen, S.M. Molecular Imaging of Metabolic Reprograming in Mutant IDH Cells. Front. Oncol. 2016, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Ryu, H.W.; Won, H.R.; Kwon, S.H. Advances in epigenetic glioblastoma therapy. Oncotarget 2017, 8, 18577–18589. [Google Scholar] [CrossRef]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Pokorny, J.L.; Cen, L.; Decker, P.A.; Wu, W.; Lomberk, G.A.; Gupta, S.K.; et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin. Cancer Res. 2012, 18, 4070–4079. [Google Scholar] [CrossRef]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef]
- Puduvalli, V.K.; Wu, J.; Yuan, Y.; Armstrong, T.S.; Vera, E.; Wu, J.; Xu, J.; Giglio, P.; Colman, H.; Walbert, T.; et al. A Bayesian adaptive randomized phase II multicenter trial of bevacizumab with or without vorinostat in adults with recurrent glioblastoma. Neuro-Oncology 2020, 22, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neuro-Oncol. 2018, 137, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncology 2018, 20, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Eessalu, T.E.; Barth, V.N.; Mitch, C.H.; Wagner, F.F.; Hong, Y.; Neelamegam, R.; Schroeder, F.A.; Holson, E.B.; Haggarty, S.J.; et al. Design, synthesis, and evaluation of hydroxamic acid-based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38. [Google Scholar]
- Gurbani, S.S.; Yoon, Y.; Weinberg, B.D.; Salgado, E.; Press, R.H.; Cordova, J.S.; Ramesh, K.K.; Liang, Z.; Velazquez Vega, J.; Voloschin, A.; et al. Assessing Treatment Response of Glioblastoma to an HDAC Inhibitor Using Whole-Brain Spectroscopic MRI. Tomography 2019, 5, 53–60. [Google Scholar] [CrossRef]
- Xu, K.; Ramesh, K.; Huang, V.; Gurbani, S.S.; Cordova, J.S.; Schreibmann, E.; Weinberg, B.D.; Sengupta, S.; Voloschin, A.D.; Holdhoff, M.; et al. Final Report on Clinical Outcomes and Tumor Recurrence Patterns of a Pilot Study Assessing Efficacy of Belinostat (PXD-101) with Chemoradiation for Newly Diagnosed Glioblastoma. Tomography 2022, 8, 688–700. [Google Scholar] [CrossRef]
- Gurbani, S.S.; Weinberg, B.D.; Salgado, E.; Voloschin, A.; Velazquez Vega, J.E.; Olson, J.J.; Shu, H.G.; Shim, H. Remarkable response of a patient with secondary glioblastoma to a histone deacetylase inhibitor. Oxf. Med. Case Rep. 2020, 2020, omaa006. [Google Scholar] [CrossRef]
- Li, B.; Yuan, M.; Kim, I.A.; Chang, C.M.; Bernhard, E.J.; Shu, H.K. Mutant epidermal growth factor receptor displays increased signaling through the phosphatidylinositol-3 kinase/AKT pathway and promotes radioresistance in cells of astrocytic origin. Oncogene 2004, 23, 4594–4602. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Cordova, J.S.; Shu, H.K.; Liang, Z.; Gurbani, S.S.; Cooper, L.A.; Holder, C.A.; Olson, J.J.; Kairdolf, B.; Schreibmann, E.; Neill, S.G.; et al. Whole-brain spectroscopic MRI biomarkers identify infiltrating margins in glioblastoma patients. Neuro-Oncology 2016, 18, 1180–1189. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Sears, T.K.; Horbinski, C.M.; Woolard, K.D. IDH1 mutant glioma is preferentially sensitive to the HDAC inhibitor panobinostat. J. Neuro-Oncol. 2021, 154, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Dow, J.; Krysztofiak, A.; Liu, Y.; Colon-Rios, D.A.; Rogers, F.A.; Glazer, P.M. Vulnerability of IDH1-Mutant Cancers to Histone Deacetylase Inhibition via Orthogonal Suppression of DNA Repair. Mol. Cancer Res. 2021, 19, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Cordova, J.S.; Kandula, S.; Gurbani, S.; Zhong, J.; Tejani, M.; Kayode, O.; Patel, K.; Prabhu, R.; Schreibmann, E.; Crocker, I.; et al. Simulating the Effect of Spectroscopic MRI as a Metric for Radiation Therapy Planning in Patients with Glioblastoma. Tomography 2016, 2, 366–373. [Google Scholar] [CrossRef]
- Ramesh, K.; Mellon, E.A.; Gurbani, S.S.; Weinberg, B.D.; Schreibmann, E.; Sheriff, S.A.; Goryawala, M.; de le Fuente, M.; Eaton, B.R.; Zhong, J.; et al. A multi-institutional pilot clinical trial of spectroscopic MRI-guided radiation dose escalation for newly diagnosed glioblastoma. Neuro-Oncol. Adv. 2022, 4, vdac006. [Google Scholar] [CrossRef]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-Oncology 2013, 15, 1114–1126. [Google Scholar] [CrossRef]
- Leather, T.; Jenkinson, M.D.; Das, K.; Poptani, H. Magnetic Resonance Spectroscopy for Detection of 2-Hydroxyglutarate as a Biomarker for IDH Mutation in Gliomas. Metabolites 2017, 7, 29. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | D-2HG (μM) | L-2HG (μM) |
|---|---|---|
| NHA | ||
| vector control | 0.07 | 0.10 |
| mtIDH1 | 18.50 | 0.06 |
| LN229 | ||
| vector control | 0.06 | 0.03 |
| mtIDH1 | 5.85 | 0.05 |
| Spiked with specific levels | ||
| 50 μM (D- or L-2HG) | 58.00 | 53.30 |
| 100 μM (D- or L-2HG) | 115.00 | 120.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-M.; Ramesh, K.K.; Huang, V.; Gurbani, S.; Kleinberg, L.R.; Weinberg, B.D.; Shim, H.; Shu, H.-K.G. Mutant Isocitrate Dehydrogenase 1 Expression Enhances Response of Gliomas to the Histone Deacetylase Inhibitor Belinostat. Tomography 2023, 9, 942-954. https://doi.org/10.3390/tomography9030077
Chang C-M, Ramesh KK, Huang V, Gurbani S, Kleinberg LR, Weinberg BD, Shim H, Shu H-KG. Mutant Isocitrate Dehydrogenase 1 Expression Enhances Response of Gliomas to the Histone Deacetylase Inhibitor Belinostat. Tomography. 2023; 9(3):942-954. https://doi.org/10.3390/tomography9030077
Chicago/Turabian StyleChang, Chi-Ming, Karthik K. Ramesh, Vicki Huang, Saumya Gurbani, Lawrence R. Kleinberg, Brent D. Weinberg, Hyunsuk Shim, and Hui-Kuo G. Shu. 2023. "Mutant Isocitrate Dehydrogenase 1 Expression Enhances Response of Gliomas to the Histone Deacetylase Inhibitor Belinostat" Tomography 9, no. 3: 942-954. https://doi.org/10.3390/tomography9030077
APA StyleChang, C.-M., Ramesh, K. K., Huang, V., Gurbani, S., Kleinberg, L. R., Weinberg, B. D., Shim, H., & Shu, H.-K. G. (2023). Mutant Isocitrate Dehydrogenase 1 Expression Enhances Response of Gliomas to the Histone Deacetylase Inhibitor Belinostat. Tomography, 9(3), 942-954. https://doi.org/10.3390/tomography9030077