
MRI CNS Atrophy Pattern and the Etiologies of Progressive Ataxias


Abstract
1. Introduction
2. Materials and Methods
2.1. Classification and Nomenclature of Progressive Ataxias
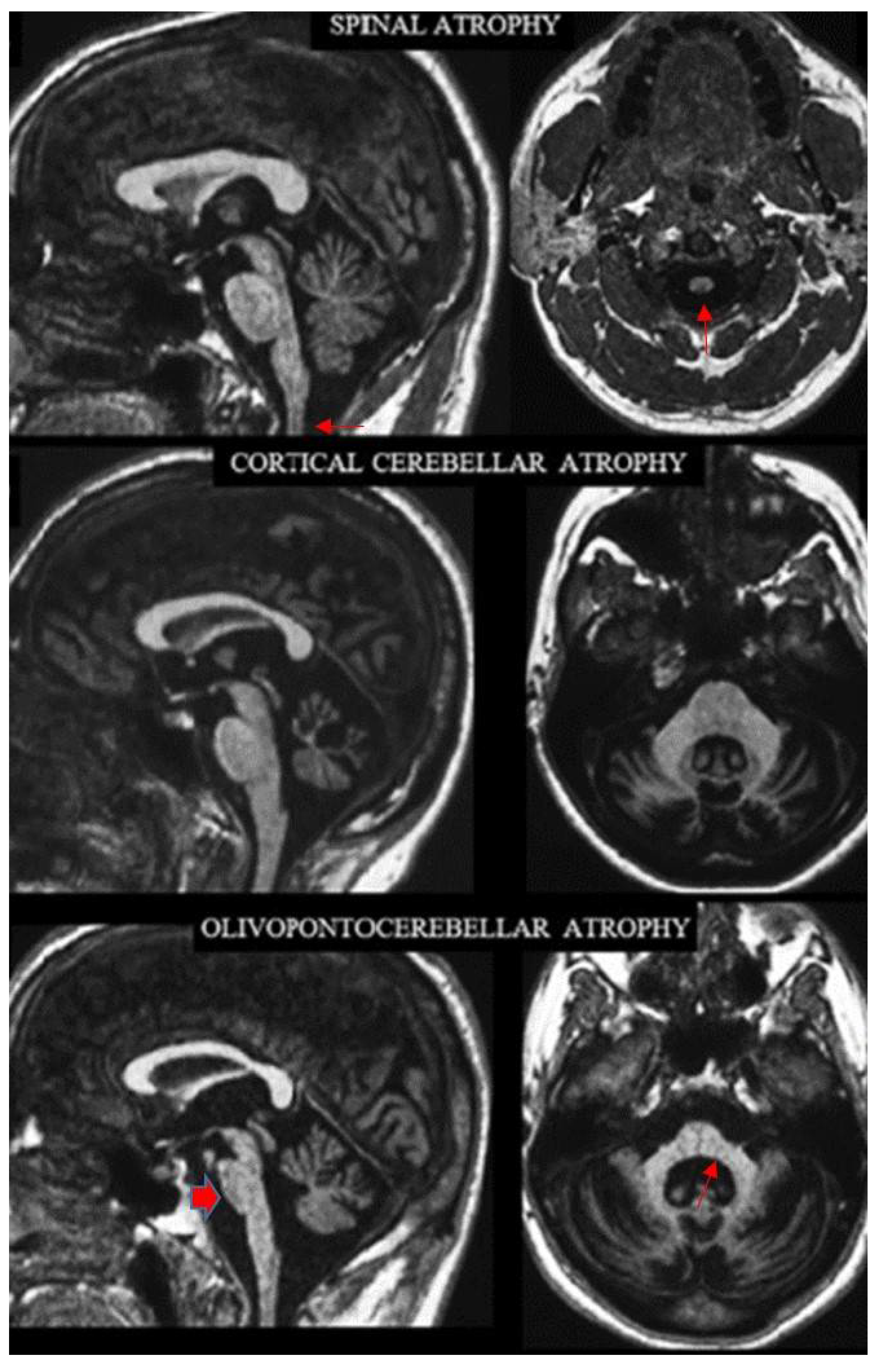
2.2. Definition of the Archetypes on MRI
2.3. Review of the MRI Features
3. Results
4. Discussion
4.1. Definite CNS Atrophy—Etiological Relationship
4.2. Pontocerebellar Hypoplasia
4.3. Uncertain CNS Atrophy—Etiological Relationship
4.4. Progressive Ataxias Characterized by MRI Signal Changes
4.5. Clinical, Diagnostic and Other Implications
4.6. Limitations
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kwon, S.; Ki, C.-S.; Youn, J.; Cho, J.W. The Etiologies of Chronic Progressive Cerebellar Ataxia in a Korean Population. J. Clin. Neurol. 2018, 14, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Friedreich, N. Ueber Ataxie mit besonderer Berucksichtigung der hereditaren Formen. Nachtrag Virchows Arch. Fu Pathol. Anat. Und Physiol. Und Fur Klin. Med. 1877, 70, 140–152. [Google Scholar] [CrossRef]
- Dejerine, J.; Thomas, A. L’atrophie olivopontocerebelleuse. Nouv. Iconogr. Salpetrière 1900, 13, 330–370. [Google Scholar]
- Marie, P.; Foix, C.; Alajouanine, T. De l’atrophie cerebelleuse tardive a predominance corticale. Rev. Neurol. 1922, 38, 1082–1111. [Google Scholar]
- Rossi, M.; Anheim, M.; Durr, A.; Klein, C.; Koenig, M.; Synofzik, M.; Marras, C.; Van De Warrenburg, B.P.; on behalf of the International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders. The genetic nomenclature of recessive cerebellar ataxias. Mov. Disord. 2018, 33, 1056–1076. [Google Scholar] [CrossRef]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- Beaudin, M.; Matilla-Dueñas, A.; Soong, B.-W.; Pedroso, J.L.; Barsottini, O.G.; Mitoma, H.; Tsuji, S.; Schmahmann, J.D.; Manto, M.; A Rouleau, G.; et al. The classification of autosomal recessive cerebellar ataxias: A consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum 2019, 18, 1098–1125. [Google Scholar] [CrossRef]
- Gebus, O.; Montaut, S.; Monga, B.; Wirth, T.; Cheraud, C.; Do Rego, C.A.; Zinchenko, I.; Carré, G.; Hamdaoui, M.; Hautecloque, G.; et al. Deciphering the causes of sporadic late-onset cerebellar ataxias: A prospective study with implications for diagnostic work. J. Neurol. 2017, 264, 1118–1126. [Google Scholar] [CrossRef]
- Futamura, N.; Matsumura, R.; Fujimoto, Y.; Horikawa, H.; Suzumura, A.; Takayanagi, T. CAG repeat expansions in patients with sporadic cerebellar ataxia. Acta Neurol. Scand. 1998, 98, 55–59. [Google Scholar] [CrossRef]
- Schöls, L.; Szymanski, S.; Peters, S.; Przuntek, H.; Epplen, J.T.; Hardt, C.; Riess, O. Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum. Genet. 2000, 107, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, R.; Tateno, F.; Kishi, M.; Tsuyusaki, Y.; Aiba, Y.; Terada, H.; Inaoka, T.; Sawai, S.; Kuwabara, S.; Nomura, F. Genetic Screening for Spinocerebellar Ataxia Genes in a Japanese Single-Hospital Cohort. J. Mov. Disord. 2017, 10, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Juvonen, V.; Hietala, M.; Kairisto, V.; Savontaus, M.L. The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol. Scand. 2005, 111, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Montaut, S.; Diedhiou, N.; Fahrer, P.; Marelli, C.; Lhermitte, B.; Robelin, L.; Vincent, M.C.; Corti, L.; Taieb, G.; Gebus, O.; et al. Biallelic RFC1-expansion in a French multicentric sporadic ataxia cohort. J. Neurol. 2021, 268, 3337–3343. [Google Scholar] [CrossRef]
- Nibbeling, E.A.R.; Duarri, A.; Verschuuren-Bemelmans, C.C.; Fokkens, M.R.; Karjalainen, J.M.; Smeets, C.J.L.M.; De Boer-Bergsma, J.J.; Van Der Vries, G.; Dooijes, D.; Bampi, G.B.; et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 2017, 140, 2860–2878. [Google Scholar] [CrossRef]
- Aydin, G.; Dekomien, G.; Hoffjan, S.; Gerding, W.M.; Epplen, J.T.; Arning, L. Frequency of SCA8, SCA10, SCA12, SCA36, FXTAS and C9orf72 repeat expansions in SCA patients negative for the most common SCA subtypes. BMC Neurol. 2018, 18, 3. [Google Scholar] [CrossRef]
- Lai, K.-L.; Liao, Y.-C.; Tsai, P.-C.; Hsiao, C.-T.; Soong, B.-W.; Lee, Y.C. Investigating PUM1 mutations in a Taiwanese cohort with cerebellar ataxia. Parkinsonism Relat. Disord. 2019, 66, 220–223. [Google Scholar] [CrossRef]
- Kim, M.; Kim, A.R.; Kim, J.S.; Park, J.; Youn, J.; Ahn, J.H.; Mun, J.K.; Lee, C.; Kim, N.-S.; Kim, N.K.; et al. Clarification of undiagnosed ataxia using whole-exome sequencing with clinical implications. Parkinsonism Relat. Disord. 2020, 80, 58–64. [Google Scholar] [CrossRef]
- Wan, N.; Chen, Z.; Wan, L.; Yuan, H.; Tang, Z.; Liu, M.; Peng, Y.; Peng, L.; Lei, L.; Xie, Y.; et al. Genetic etiology of a Chinese ataxia cohort: Expanding the mutational spectrum of hereditary ataxias. Parkinsonism Relat. Disord. 2021, 89, 120–127. [Google Scholar] [CrossRef]
- Pyle, A.; Smertenko, T.; Bargiela, D.; Griffin, H.R.; Duff, J.; Appleton, M.; Douroudis, K.; Pfeffer, G.; Santibanez-Koref, M.; Eglon, G.; et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015, 138, 76–83. [Google Scholar] [CrossRef]
- Filho, F.M.R.; Palma, M.M.; Pedroso, J.L.; Barsottini, O.G.; Sallum, J.M. PRPS1 Gene Mutation Causes Complex X-Linked Adult-Onset Cerebellar Ataxia in Women. Neurol. Genet. 2021, 7, e563. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Foroud, T.M. Neurology individualized medicine: When to use next-generation sequencing panels. Mayo Clin. Proc. 2017, 92, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Bahlo, M.; Bennett, M.F.; Degorski, P.; Tankard, R.M.; Delatycki, M.B.; Lockhart, P.J. Recent advances in the detection of repeat expansions with short-read next-generation sequencing. F1000Res 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Renaud, M.; Tranchant, C.; Martin, J.V.T.; Mochel, F.; Synofzik, M.; Van De Warrenburg, B.; Pandolfo, M.; Koenig, M.; Kolb, S.A.; Anheim, M.; et al. A recessive ataxia diagnosis algorithm for the next generation sequencing era. Ann. Neurol. 2017, 82, 892–899. [Google Scholar] [CrossRef] [PubMed]
- van de Warrenburg, B.P.; van Gaalen, J.; Ginestroni, A.; Mascalchi, M. Acute and Subacute Ataxia. In Imaging Acute Neurologic Disease. A Symptom-Based Approach; Filippi, M., Simon, J.H., Eds.; Cambridge University Press: Cambridge, UK, 2014; pp. 144–165. [Google Scholar]
- Wüllner, U.; Klockgether, T.; Petersen, D.; Naegele, T.; Dichgans, J. Magnetic resonance imaging in hereditary and idiopathic ataxia. Neurology 1993, 43, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, I.E.; Harding, A.E.; Miller, D.H.; Johnson, G.; MacManus, D.; du Boulay, E.P.; Kendall, B.E.; Moseley, I.F.; McDonald, W.I. Magnetic resonance imaging in degenerative ataxic disorders. J. Neurol. Neurosurg. Psychiatry 1994, 57, 51–57. [Google Scholar] [CrossRef][Green Version]
- Mascalchi, M. Spinocerebellar ataxias. Neurol. Sci. 2008, 29, 311–313. [Google Scholar] [CrossRef]
- Mascalchi, M.; Vella, A. Neuroimaging Applications in Chronic Ataxias. Int. Rev. Neurobiol. 2018, 143, 109–162. [Google Scholar] [CrossRef]
- Heidelberg, D.; Ronsin, S.; Bonneville, F.; Hannoun, S.; Tilikete, C.; Cotton, F. Main inherited neurodegenerative cerebellar ataxias, how to recognize them using magnetic resonance imaging? J. Neuroradiol. 2018, 45, 265–275. [Google Scholar] [CrossRef]
- Lieto, M.; Roca, A.; Santorelli, F.M.; Fico, T.; De Michele, G.; Bellofatto, M.; Saccà, F.; De Michele, G.; Filla, A. Degenerative and acquired sporadic adult onset ataxia. Neurol. Sci. 2019, 40, 1335–1342. [Google Scholar] [CrossRef]
- De Silva, R.N.; Vallortigara, J.; Greenfield, J.; Hunt, B.; Giunti, P.; Hadjivassiliou, M. Diagnosis and management of progressive ataxia in adults. Pract. Neurol. 2019, 19, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Pontillo, G.; De Michele, G.; Di Stasi, M.; Guerriero, E.; Perillo, T.; Pane, C.; De Rosa, A.; Ugga, L.; Brunetti, A. Conventional MRI findings in hereditary degenerative ataxias: A pictorial review. Neuroradiology 2021, 63, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Mascalchi, M. Conventional MRI features in progressive ataxias. Splitting or clumping? Neuroradiology 2020, 63, 1745–1746. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.H. Ataxia. Continuum 2019, 25, 1036–1054. [Google Scholar] [CrossRef] [PubMed]
- Poretti, A.; Boltshauser, E. Terminology in morphological anomalies of the cerebellum does matter. Cerebellum Ataxias 2015, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Savoiardo, M.; Strada, L.; Girotti, F.; A Zimmerman, R.; Grisoli, M.; Testa, D.; Petrillo, R. Olivopontocerebellar atrophy: MR diagnosis and relationship to multisystem atrophy. Radiology 1990, 174, 693–696. [Google Scholar] [CrossRef]
- Namavar, Y.; Barth, P.G.; Kasher, P.; Van Ruissen, F.; Brockmann, K.; Bernert, G.; Writzl, K.; Ventura, K.; Cheng, E.Y.; Ferriero, D.M.; et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 2011, 134, 143–156. [Google Scholar] [CrossRef]
- Mascalchi, M.; Salvi, F.; Piacentini, S.; Bartolozzi, C. Friedreich’s ataxia: MR findings involving the cervical portion of the spinal cord. AJR Am. J. Roentgenol. 1994, 163, 187–191. [Google Scholar] [CrossRef]
- Mascalchi, M.; Cosottini, M.; Lolli, F.; Salvi, F.; Tessa, C.; Macucci, M.; Tosetti, M.; Plasmati, R.; Ferlini, A.; Tassinari, C.A.; et al. Proton MR spectroscopy of the cerebellum and pons in patients with degenerative ataxia. Radiology 2002, 223, 371–378. [Google Scholar] [CrossRef]
- Shanmugarajah, P.D.; Hoggard, N.; Currie, S.; Aeschlimann, D.P.; Aeschlimann, P.C.; Gleeson, D.C.; Karajeh, M.; Woodroofe, N.; Grünewald, R.A.; Hadjivassiliou, M. Alcohol-related cerebellar degeneration: Not all down to toxicity? Cerebellum Ataxias 2016, 3, 17. [Google Scholar] [CrossRef]
- Breza, M.; Bourinaris, T.; Efthymiou, S.; Maroofian, R.; Athanasiou-Fragkouli, A.; Tzartos, J.; Velonakis, G.; Karavasilis, E.; Angelopoulou, G.; Kasselimis, D.; et al. A homozygous GDAP2 loss-of-function variant in a patient with adult-onset cerebellar ataxia. Brain 2020, 143, e49. [Google Scholar] [CrossRef]
- Beecroft, S.J.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dyer, Z.; Wu, T.Y.; Mulroy, E.; Pelosi, L.; Rodrigues, M.; Taylor, R.; et al. A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 2020, 143, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Tozza, S.; Yau, W.Y.; Rossi, S.; Beecroft, S.J.; Jaunmuktane, Z.; Dyer, Z.; Ravenscroft, G.; Lamont, P.J.; Mossman, S.; et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 2020, 143, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Chen, Z.; Wan, N.; Liu, M.; Xue, J.; Chen, H.; Zhang, Y.; Peng, Y.; Tang, Z.; Gong, Y.; et al. Biallelic Intronic AAGGG Expansion of RFC1 is Related to Multiple System Atrophy. Ann. Neurol. 2020, 88, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Traschütz, A.; Cortese, A.; Reich, S.; Dominik, N.; Faber, J.; Jacobi, H.; Hartmann, A.M.; Rujescu, D.; Montaut, S.; Echaniz-Laguna, A.; et al. Natural History, Phenotypic Spectrum, and Discriminative Features of Multisystemic RFC1 Disease. Neurology 2021, 96, e1369–e1382. [Google Scholar] [CrossRef] [PubMed]
- Hattingen, E.; Pilatus, U. (Eds.) Brain Tumor Imaging; Springer: Berlin/Heidelberg, Germany, 2016; ISSN 0942-3573. [Google Scholar]
- Kumar, N.; Pandey, S.; Garg, R.K.; Holla, V.V. A case of subacute progressive cerebellar ataxia: Brownell-Oppenheimer variant of sporadic Creutzfeldt-Jakob disease. Neurol. India 2021, 69, 217–218. [Google Scholar] [CrossRef]
- Chen, D.-H.; Below, J.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef]
- Knight, M.A.; Gardner, R.J.M.; Bahlo, M.; Matsuura, T.; Dixon, J.A.; Forrest, S.M.; Storey, E. Dominantly inherited ataxia and dysphonia with dentate calcification: Spinocerebellar ataxia type 20. Brain 2004, 127, 1172–1181. [Google Scholar] [CrossRef]
- Dotti, M.T.; Federico, A.; Signorini, E.; Caputo, N.; Venturi, C.; Filosomi, G.; Guazzi, G.C. Cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease): CT and MR findings. AJNR Am. J. Neuroradiol. 1994, 15, 1721–1726. [Google Scholar]
- Seijo-Martínez, M.; Navarro, C.; Del Río, M.C.; Vila, O.; Puig, M.; Ribes, A.; Butron, M. L-2-hydroxyglutaric aciduria: Clinical, neuroimaging, and neuropathological findings. Arch. Neurol. 2005, 62, 666–670. [Google Scholar] [CrossRef]
- Biancheri, R.; Rosano, C.; Denegri, L.; Lamantea, E.; Pinto, F.; Lanza, F.; Severino, M.; Filocamo, M. Expanded spectrum of Pelizaeus-Merzbacher-like disease: Literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur. J. Hum. Genet. 2013, 21, 34–39. [Google Scholar] [CrossRef][Green Version]
- Magen, D.; Georgopoulos, C.; Bross, P.; Ang, D.; Segev, Y.; Goldsher, D.; Nemirovski, A.; Shahar, E.; Ravid, S.; Luder, A.; et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008, 83, 30–42. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Van Der Voorn, P.; Barkhof, F.; Van Coster, R.; Krägeloh-Mann, I.; Feigenbaum, A.; Blaser, S.; Vles, J.S.H.; Rieckmann, P.; Pouwels, P.J.W. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann. Neurol. 2003, 53, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Bugiani, M.; Dupuits, C.; Galanaud, D.; Touitou, V.; Postma, N.; van Berkel, C.; Polder, E.; Tollard, E.; Darios, F.; et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: An observational analytical study. Lancet Neurol. 2013, 12, 659–668. [Google Scholar] [CrossRef]
- Rantamaki, M.; Krahe, R.; Paetau, A.; Cormand, B.; Mononen, I.; Udd, B. Adult-onset autosomal recessive ataxia with thalamic lesions in a Finnish family. Neurology 2001, 57, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Meijer, I.A.; Lessel, D.; Mencacci, N.E.; Krainc, D.; Hempel, M.; Tsiakas, K.; Prokisch, H.; Rossignol, E.; Helm, M.H.; et al. Recessive mutations in VPS13D cause childhood onset movement disorders. Ann. Neurol. 2018, 83, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Apartis, E.; Blancher, A.; Meissner, W.G.; Guyant-Maréchal, L.; Maltête, D.; De Broucker, T.; Legrand, A.-P.; Bouzenada, H.; Thanh, H.T.; Sallansonnet-Froment, M.; et al. FXTAS: New insights and the need for revised diagnostic criteria. Neurology 2012, 79, 1898–1907. [Google Scholar] [CrossRef]
- De Michele, G.; Filla, A. Other autosomal recessive and childhood ataxias. Handb. Clin. Neurol. 2012, 103, 343–357. [Google Scholar] [CrossRef]
- Klockgether, T.; Petersen, D.; Grodd, W.; Dichgans, J. Early onset cerebellar ataxia with retained tendon reflexes. Clinical, electrophysiological and MRI observations in comparison with Friedreich’s ataxia. Brain 1991, 114, 1559–1573. [Google Scholar] [CrossRef]
- De Michele, G.; Di Salle, F.; Filla, A.; D’Alessio, A.; Ambrosio, G.; Viscardi, L.; Scala, R.; Campanella, G. Magnetic resonance imaging in “typical” and “late onset” Friedreich’s disease and early onset cerebellar ataxia with retained tendon reflexes. Ital. J. Neurol. Sci. 1995, 16, 303–308. [Google Scholar] [CrossRef]
- Mariotti, C.; Gellera, C.; Rimoldi, M.; Mineri, R.; Uziel, G.; Zorzi, G.; Pareyson, D.; Piccolo, G.; Gambi, D.; Piacentini, S.; et al. Ataxia with isolated vitamin E deficiency: Neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol. Sci. 2004, 25, 130–137. [Google Scholar] [CrossRef] [PubMed]
- El Euch-Fayache, G.; Bouhlal, Y.; Amouri, R.; Feki, M.; Hentati, F. Molecular, clinical and peripheral neuropathy study of Tunisian patients with ataxia with vitamin E deficiency. Brain 2014, 137, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Eidhof, I.; Baets, J.; Kamsteeg, E.-J.; Deconinck, T.; Van Ninhuijs, L.; Martin, J.-J.; Schüle, R.; Züchner, S.; De Jonghe, P.; Schenck, A.; et al. GDAP2 mutations implicate susceptibility to cellular stress in a new form of cerebellar ataxia. Brain 2018, 141, 2592–2604. [Google Scholar] [CrossRef] [PubMed]
- Syriani, D.A.; Wong, D.; Andani, S.; De Gusmao, C.M.; Mao, Y.; Sanyoura, M.; Glotzer, G.; Lockhart, P.J.; Hassin-Baer, S.; Khurana, V.; et al. Prevalence of RFC1-mediated spinocerebellar ataxia in a North American ataxia cohort. Neurol. Genet. 2020, 6, e440. [Google Scholar] [CrossRef]
- Subramony, S.; Advincula, J.; Perlman, S.; Rosales, R.L.; Lee, L.V.; Ashizawa, T.; Waters, M.F. Comprehensive phenotype of the p.Arg420his allelic form of spinocerebellar ataxia type 13. Cerebellum 2013, 12, 932–936. [Google Scholar] [CrossRef]
- Klockgether, T. The clinical diagnosis of autosomal dominant spinocerebellar ataxias. Cerebellum 2008, 7, 101–105. [Google Scholar] [CrossRef]
- Harding, I.H.; Chopra, S.; Arrigoni, F.; Boesch, S.; Brunetti, A.; Cocozza, S.; Corben, L.A.; Deistung, A.; Delatycki, M.; Diciotti, S.; et al. Brain Structure and Degeneration Staging in Friedreich Ataxia: Magnetic Resonance Imaging Volumetrics from the ENIGMA-Ataxia Working Group. Ann. Neurol. 2021, 90, 570–583. [Google Scholar] [CrossRef]
- Martins, C.R., Jr.; Martinez, A.R.M.; De Rezende, T.J.R.; Branco, L.M.T.; Pedroso, J.L.; Barsottini, O.G.P.; Lopes-Cendes, I.; França, M.C. Spinal cord damage in Spinocerebellar Ataxia type 1. Cerebellum 2017, 16, 792–796. [Google Scholar] [CrossRef]
- Fahl, C.N.; Branco, L.M.T.; Bergo, F.P.G.; D’Abreu, A.; Lopes-Cendes, I.; França, M.C., Jr. Spinal cord damage in Machado-Joseph disease. Cerebellum 2015, 14, 128–132. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ohta, Y.; Kobayashi, H.; Okamoto, M.; Takamatsu, K.; Ota, T.; Manabe, Y.; Okamoto, K.; Koizumi, A.; Abe, K. Clinical features of SCA36: A novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology 2012, 79, 333–341. [Google Scholar] [CrossRef]
- Herman-Bert, A.; Stevanin, G.; Netter, J.-C.; Rascol, O.; Brassat, D.; Calvas, P.; Camuzat, A.; Yuan, Q.-P.; Schalling, M.; Dürr, A.; et al. Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am. J. Hum. Genet. 2000, 67, 229–235. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jacobi, H.; Reetz, K.; du Montcel, S.T.; Bauer, P.; Mariotti, C.; Nanetti, L.; Rakowicz, M.; Sulek, A.; Durr, A.; Charles, P.; et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: Analysis of baseline data. Lancet Neurol. 2013, 12, 650–658. [Google Scholar] [CrossRef]
- Genis, D.; Ortega-Cubero, S.; Nicolás, H.S.; Corral, J.; Gardenyes, J.; de Jorge, L.; López, E.; Campos, B.; Lorenzo, E.; Tonda, R.; et al. Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 2018, 91, e1988–e1998. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Kumamoto, T.; Masuda, T.; Nomura, Y.; Hanaoka, T.; Hazama, Y.; Okazaki, T.; Arakawa, R. Evaluation of the effect of thyrotropin releasing hormone (TRH) on regional cerebral blood flow in spinocerebellar degeneration using 3DSRT. J. Neurol. Sci. 2009, 281, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Pontillo, G.; De Michele, G.; Perillo, T.; Guerriero, E.; Ugga, L.; Salvatore, E.; Galatolo, D.; Riso, V.; Saccà, F.; et al. The “crab sign”: An imaging feature of spinocerebellar ataxia type 48. Neuroradiology 2020, 62, 1095–1103. [Google Scholar] [CrossRef]
- Della Nave, R.; Foresti, S.; Tessa, C.; Moretti, M.; Ginestroni, A.; Gavazzi, C.; Guerrini, L.; Salvi, F.; Piacentini, S.; Mascalchi, M. ADC mapping of neurodegeneration in the brainstem and cerebellum of patients with progressive ataxias. Neuroimage 2004, 22, 698–705. [Google Scholar] [CrossRef]
- Della Nave, R.; Ginestroni, A.; Tessa, C.; Salvatore, E.; Bartolomei, I.; Salvi, F.; Dotti, M.T.; De Michele, G.; Piacentini, S.; Mascalchi, M. Brain white matter tracts degeneration in Friedreich ataxia. An in vivo MRI study using tract-based spatial statistics and voxel-based morphometry. Neuroimage 2008, 40, 19–25. [Google Scholar] [CrossRef]
- Della Nave, R.; Ginestroni, A.; Tessa, C.; Salvatore, E.; De Grandis, D.; Plasmati, R.; Salvi, F.; De Michele, G.; Dotti, M.T.; Piacentini, S.; et al. Brain white matter damage in SCA1 and SCA2. An in vivo study using voxel-based morphometry, histogram analysis of mean diffusivity and tract-based spatial statistics. Neuroimage 2008, 43, 10–19. [Google Scholar] [CrossRef]
- Bonilha Da Silva, C.; Bergo, F.P.G.; D’Abreu, A.; Cendes, F.; Lopes-Cendes, I.; França, M.C., Jr. Dentate nuclei T2 relaxometry is a reliable neuroimaging marker in Friedreich’s ataxia. Eur. J. Neurol. 2014, 21, 1131–1136. [Google Scholar] [CrossRef]
- Ronsin, S.; Hannoun, S.; Thobois, S.; Petiot, P.; Vighetto, A.; Cotton, F.; Tilikete, C. A new MRI marker of ataxia with oculomotor apraxia. Eur. J. Radiol. 2019, 110, 187–192. [Google Scholar] [CrossRef]


{kind=link}
| MRI CNS Atrophy Pattern | Etiology | ||
|---|---|---|---|
| Spinal Atrophy (SA) | Cerebellar Cortical Atrophy (CCA) | Olivopontocerebellar Atrophy (OPCA) | |
| Alcoholic cerebellar degeneration Gluten ataxia Anti-GAD ataxia Paraneoplastic cerebellar degeneration SAOA/ILOCA | MSA-C | Acquired | |
| - | SCA5(600224), SCA6(183086) SCA8(608768), SCA10(603516) SCA11(604432), SCA12(604326) SCA13(605259), SCA14(605361) SCA15/16(606658) SCA19/22(607346), SCA20(608687) SCA21(607454), SCA26(609306), SCA27(609307) SCA28(610246), SCA31(117210) SCA35(613908), SCA37(615945) SCA38(615957), SCA42(616795) SCA43(617018) SCA44(617691), SCA47(617931) SCA48(618093) | DRPLA(125370) SCA1(164400), SCA2(183090) SCA3(109150), SCA7(164500) SCA23(610245), SCA29(117360), SCA34(133190), SCA36(614153) | Inherited Dominant |
| FRDA (229300) | AOA1 (208920) AOA2/SCAN2/SCAR1 (606002) AOA4(616267) ARCA1/SCAR8 (610743) ARCA2(612016) ARSACS (270550) AT (208900) Cayman ataxia(601238) CLN11(614706) GHS(212840) Gillespie syndrome(206700) HLD7(607694), HLD8(614381) Mitochondrial complex IV deficiency(220110) MTDP7 syndrome(271245) MGCA5(610198) MMS(248800) PBD6B(614871) PHARC(612674) Progressive myoclonic epilepsy 6(614018) SCAN1(607250), SCAN3(618387) SCAR2(213200) SCAR5(25130) SCAR9(612016), SCAR10(613728) SCAR11(614229) SCAR13(614831), SCAR14(615386) SCAR16(615768), SCAR17(616127) SCAR18(616204), SCAR19(616291) SCAR20(616354), SCAR21(616719) SCAR22(616948), SCAR24(617133) SCAR25(617584), SCAR26(617633) SCAR28 (618800), SCAR29(619389) SCAR30(619405), SCAR31(619422) SPAX5(614487) SPG7 (607259), SPG46(614409) SPG76(616907), SPG79(615491) | BNS (215470/2754) SCAR7(609270) | Inherited Recessive |
| - | SCAX1(302500) | PRPS1 Gene Mutation | Inherited X-linked |
| Etiology | Signal Changes | References Number | |
|---|---|---|---|
| Acquired | Tumors | Variable | [47] |
| Kreutzfeld–Jakob disease | Basal ganglia and cortex hyperintensity in T2-weighted images and decreased diffusion | [48] | |
| Siderosis | Low signal rim of CNS surfaces in T2 or T2*-weighted images | [31] | |
| Vit B12 deficiency | Hyperintense posterior columns of the spinal cord in T2-weighted images | [31] | |
| Dominantly inherited | ATXPC(159550) | Hyperintense cerebral WM in T2-weighted images | [49] |
| SCA20 (608687) | Low signal of the dentate due to calcifications in T2 or T2*—weighted images | [50] | |
| Recessively inherited | CTX(213700) | Hyperintense peridentate and cerebral WM in T2-weighted images | [51] |
| 2-Hydroxic Glutaric Aciduria (236792) | Hyperintense cerebral WM in T2-weighted images | [52] | |
| HLD2(608804) | Hyperintense cerebral, cerebellar, brainstem and spinal cord WM in T2-weighted images | [53] | |
| HLD4(612233) | Lack of normal WM myelination | [54] | |
| LBSL(611105) | Hyperintense cerebral WM and WM tracts in cerebral and cerebellar peduncles, brainstem and spinal cord in T2-weighted images | [55] | |
| Leukoencephalopthy with ataxia (615651) | Hyperintense internal capsule, cerebral peduncles and middle cerebellar peduncles in T2-weighted images with restricted diffusion | [56] | |
| SANDO(607459) | Hyperintense thalami, middle cerebellar peduncle and cerebellar WM in T2-weighted images | [57] | |
| SCAR4/SCA24(607317) | Hyperintense basal ganglia and cerebral WM in T2-weighted images | [58] | |
| X-linked inherited | FXTAS (300623) | Hyperintensity of the middle cerebellar peduncles, splenium corpus callosum and of the cerebral WM in T2-weighted images | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascalchi, M. MRI CNS Atrophy Pattern and the Etiologies of Progressive Ataxias. Tomography 2022, 8, 423-437. https://doi.org/10.3390/tomography8010035
Mascalchi M. MRI CNS Atrophy Pattern and the Etiologies of Progressive Ataxias. Tomography. 2022; 8(1):423-437. https://doi.org/10.3390/tomography8010035
Chicago/Turabian StyleMascalchi, Mario. 2022. "MRI CNS Atrophy Pattern and the Etiologies of Progressive Ataxias" Tomography 8, no. 1: 423-437. https://doi.org/10.3390/tomography8010035
APA StyleMascalchi, M. (2022). MRI CNS Atrophy Pattern and the Etiologies of Progressive Ataxias. Tomography, 8(1), 423-437. https://doi.org/10.3390/tomography8010035