1. Introduction
Blood coagulation is a dynamic process of changing blood from a liquid state to a non-flowing gel state, which is an important part of physiological hemostasis. The essence of blood coagulation is the process by which soluble fibrinogen in blood plasma becomes insoluble fibrin [
1,
2]. The blood coagulation cascade is a sequence of physiological reactions that culminate in the formation of thrombin. This process involves multiple steps, many of which require the presence of non-enzymatic cofactors [
3]. These cofactors are essential for the cascade’s proper function and are categorized into two main classes: clotting factors and negatively charged surfaces [
4]. Phosphatidylserine (PS) exposure on platelet membranes is central to the regulation of blood coagulation and thrombin production [
5]. It is widely accepted that the negatively charged surface of PS-containing platelet-derived membranes contributes significantly to this rate enhancement. However, the precise mechanism through which this occurs is still debated [
6].
Recent advances in viscoelastic assays, such as thromboelastography and rotational thromboelastometry, provide a more comprehensive assessment of coagulation by continuously monitoring clot formation and breakdown. However, conventional screening tests like prothrombin time (PT) and activated partial thromboplastin time (APTT) remain widely used as the standard laboratory tests for evaluating coagulation defects [
7,
8]. While PT and APTT are automated and convenient, they have significant limitations as static in vitro assays that use crude activators and cannot replicate the intricate processes of coagulation activation and propagation in vivo [
9]. Specifically, the APTT test utilizes a variety of commercial reagents consisting of different contact activators, phospholipid sources, and concentrations, leading to substantial variability in results [
10,
11,
12,
13,
14]. For instance, the use of different platelet-derived phospholipids results in poor consistency of APTT clotting times, which hampers clinical interpretation and decision making on the bleeding risks of patients. Moreover, the clotting times derived from these reagents show poor correlation with thrombin generation profiles in patient plasma. Therefore, the APTT assay may fail to identify some coagulation abnormalities. To address the limitations of existing reagents and more accurately identify coagulopathies, it is imperative to develop novel APTT reagents with standardized phospholipid compositions and activators that can closely mimic physiological coagulation. In this study, we aim to formulate and validate new APTT reagents with optimized phospholipids and activators to minimize assay variability and establish reference ranges that better guide clinical diagnosis and management of coagulation disorders.
To overcome the limitations of conventional APTT reagents, the purpose of this study was to investigate the procoagulant effect of apoptotic cell-inspired polymeric particles as a biomimetic material to affect the blood coagulation system. In our previous study, apoptotic cell membrane-inspired methacryloyloxyethyl phosphorylserine (MPS) particles were helpful for immunosuppressive and post inflammatory effects on macrophages [
15]. Phosphatidylserine (PS) is known for its anti-inflammatory effects as well as its role in blood coagulation. In our previous research, we demonstrated that synthetic MPS particles exhibit similar anti-inflammatory effects as natural PS, effectively modulating immune responses in macrophages. Due to these physiological functions, it is expected that these MPS particles will also influence the blood coagulation process. Herein, biomimetic MPS particles are proposed in this study to approach the commercial APTT reagent for comparing procoagulant activity.
2. Materials and Methods
2.1. Materials
Butyl methacrylate (BMA), hydroxyethyl methacrylate (HEMA), 2,2′-azobis(isobutyronitrile) (AIBN), and imidazole hydrochloride were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). O-tert-butoxy-N,N,N,N,-tetraisopropyl phosphoramidite, tert-butyl peroxide, aluminum chloride hexahydrate, calcium chloride, ellagic acid, and glutaraldehyde solution, 25% in H2O, were purchased from Sigma-Aldrich (St. Louis, MO, USA). Trifluoroacetic acid (TFA) and chloroform-d were purchased from Tokyo Chemical Industry (Tokyo, Japan). N-Boc-l-serine tert-butyl ester was purchased from Watanabe Chemical Industries (Hiroshima, Japan). Phosphate-buffered saline and sodium hydroxide were purchased from Nacalai tesque (Kyoto, Japan). Coagtrol N and Actin FSL kits were purchased from Sysmex Corporation (Kobe, Japan). N-2-hydeoxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES) buffer solution was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Other reagents and solvents are commercial extra-pure grade and were used without further purification.
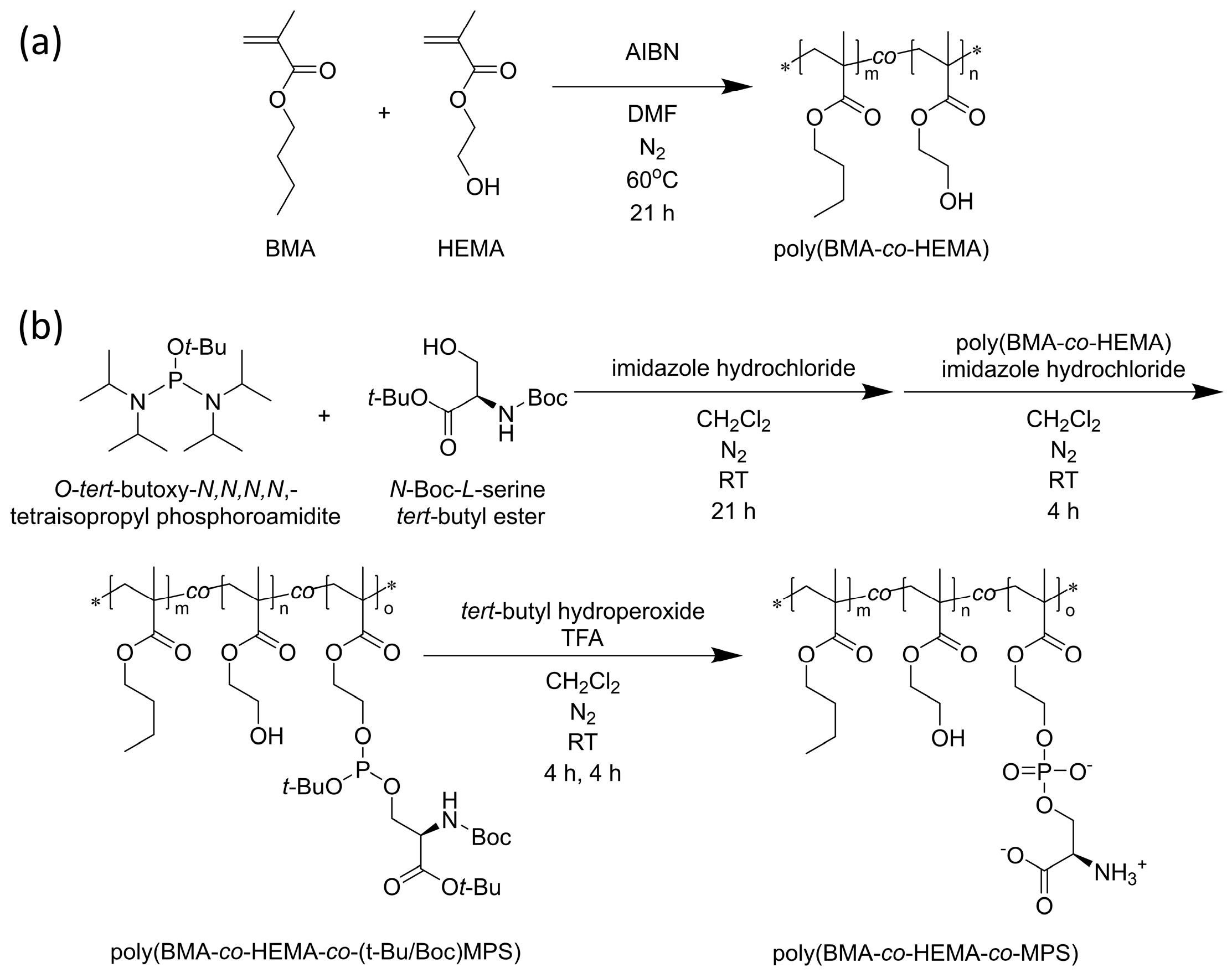
2.2. Synthesis of Poly (BMA-co-HEMA-co-MPS)
A random copolymer of BMA, HEMA, and MPS was synthesized by previous methods [
14,
15,
16]. Briefly, poly (BMA-
co-HEMA) was polymerized by free radical polymerization with 7.5 mmol of BMA, 35 mmol of HEMA, and 0.04 mmol of AIBN in 40 mL of DMF at 60 °C for 21 h (
Scheme 1a).
Subsequently, the hydroxyl group of serine, whose amine and carboxyl groups are protected, was modified to the side chain hydroxyl group of the resulting poly (BMA-co-HEMA) using a phosphoramidite reagent, a phosphate ester-forming reagent. Specifically, 9 mmol of O-tert-butoxy-N,N,N,N,-tetraisopropyl phosphoramidite, 10 mmol of N-Boc-l-serine tert-butyl ester, and 2.2 mmol of imidazole hydrochloride were stirred in 200 mL of super-dehydrated dichloromethane in a N2 atmosphere for 21 h at room temperature. poly (BMA-co-HEMA) was placed in the system, and a total of 28 mmol of imidazole hydrochloride was added in three equal portions that were spaced out by 45 min each under N2 atmosphere. At 150 min after the last addition of imidazole hydrochloride, the mixture was purified by dialysis against 2-propanol four times and against dichloromethane twice at 4 °C using a cellulose dialysis membrane (MWCO = 1000). A small part of the solution was measured by 1H-NMR spectroscopy to evaluate the modification degree of the PS group.
To the resulting mixture, 22 mmol of
tert-butyl peroxide was added. The mixture was stirred at room temperature for 6 h, followed by purification through dialysis against dichloromethane twice at 4 °C (MWCO = 1000). Next, trifluoroacetic acid was added to the solution, which was then stirred at room temperature for another 6 h. The mixture was further purified by dialysis against a 0.1 M NaOH solution twice, followed by isopropanol twice, and methanol twice, all at 4 °C (MWCO = 1000). Finally, poly(BMA-
co-HEMA-
co-MPS) was obtained by removing the solvent using a rotary evaporator and a vacuum pump overnight (
Scheme 1b).
The modification of the PS group was confirmed by measuring the FT-IR spectrum to detect the stretching vibration of PO2− in the polymer.
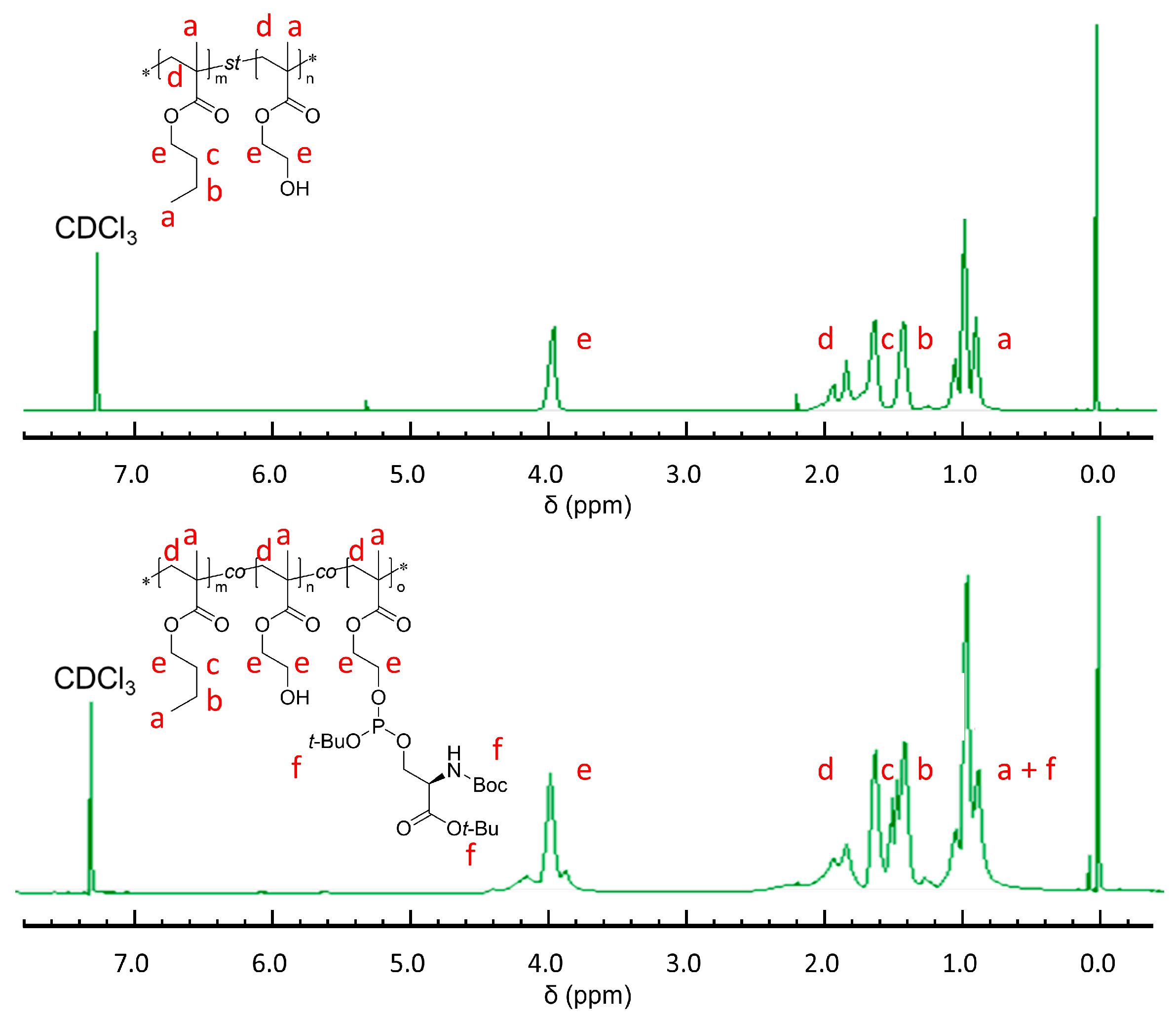
1H-NMR of poly (BMA-co-HEMA): (CDCl3-d, 300 MHz): δ = 0.80–1.10 (-CH3, -CH3 in the chain, broad, (6m + 3n)H), δ = 1.40 (-CH2-CH3, s, 2nH), δ = 1.60 (-CH2-CH2- CH3, s, 2mH), δ = 1.80–2.00 (-CH2-C(CH3)(COO-)CH2- in the chain, broad, 2(m + n)H), δ = 3.80 (-CH2-OH, s, 2nH), δ = 4.00 (-COO-CH2- in the BMA site, s, 2mH), δ = 4.10 (-COO-CH2- in the HEMA site, s, 2nH). GPC: Mn = 12.9 × 103 g mol−1, Mw/Mn = 1.13.
1H-NMR of poly (BMA-co-HEMA-co-MPS) before deprotection: δ = 0.80–1.10 (-CH3, -CH3 in the chain, broad, (6m + 3n + 3o)H), δ = 1.30–1.70 (-CH2-CH3, s, (2n + 2o)H; -CH2-CH2-CH3, s, 2mH; tert-Bu group, multi, 27oH), δ = 1.80–2.40 (-CH2-C(CH3)(COO-)CH2- in the chain, broad, 2(m + n + o)H), δ = 3.80–4.10 (-CH2-OH, s, 2nH); (-COO-CH2- in the BMA site, s, 2mH), δ = (-COO-CH2- in the HEMA and PMS site, s, 2(n + o)H). GPC: Mn = 8.30 × 103 g mol−1, Mw/Mn = 1.18.
2.3. Preparation of PS Particles of Poly (BMA-co-HEMA-co-MPS)
The PS particle was prepared through a self-assembly mechanism adopting simple dialysis following the previous method with small modifications [
7,
9]. Poly (BMA-
co-HEMA-
co-MPS) was dissolved in three different concentrations at 2 mg/mL, 4 mg/mL, and 8 mg/mL in DMF, and solutions were dialyzed against distilled water for 3 days at 4 °C using a dialysis membrane (MWCO = 1000). The water was replaced every 12 h. Finally, the sample solution in the dialysis membrane was collected into a glass vial and freeze dried for 3 days to achieve a white powder.
2.4. Morphology, Size, and Zeta Potential of PS Particles
Sonification was applied to the three different diluted PS particles in DI water (1 mg/mL) in a water bath desktop ultrasonic cleaner US100 series (SANSYO Co., Ltd., Company, Tokyo, Japan) for about 30 min. Before the operation, the measurement duration was set to be determined automatically. The morphology, size, and zeta potential of PS particles were evaluated by field emission-scanning electron microscopy (FESEM; Hitachi S-4700 I, Tokyo, Japan) and a zeta potential laser/particle analyzer (Dynamic light scattering (DLS), Otsuka Electronics Co., Ltd., Osaka, Japan), respectively (Figures 3 and 4).
The SEM samples were prepared by being mounted on freeze-dried samples to aluminum SEM pins and coated with Au/Pd using a sputter coating instrument provided at 20 mA for 120 s (Figure 6).
The zeta potentials of PS particles (prepared at a concentration of 2 mg/mL) were evaluated at different concentrations of 0.0006 to 0.6 mg/mL by following the above procedure (Figure 7).
All of the measurements were performed at room temperature. The software provided by the manufacturer was used to automatically calculate the size of the particles and the polydispersity index. The diameter mean values were calculated from the measurements performed at least in triplicate.
2.5. Evaluation of Procoagulant Efficiency of PS Particles by APTT Assay
The influence of particle size and concentration on blood coagulation was evaluated using the Activated Partial Thromboplastin Time (APTT) assay. Coagtrol N was prepared by dissolving it in 1 mL of deionized (DI) water and gently overturning for 30 min at room temperature to ensure stable reagents and remove the bubbles. Ellagic acid was dissolved in a 50 mM HEPES buffer solution (pH 7.35) to a final concentration of 0.1 mM and mixed with 50 μM aluminum chloride hexahydrate as an antioxidant.
To assess the size-dependent effects on coagulation, PS particles of three different sizes (907 nm, 937 nm, and 976 nm) at a fixed concentration of 0.06 mg/mL were tested (Figure 4). Additionally, the influence of particle concentration was explored using PS particle solutions at varying concentrations (0.0006, 0.006, 0.06, 0.6, 2, 4, and 8 mg/mL) (Figure 5). For both sets of experiments, 50 μL of each PS particle solution or concentration variant was mixed with 50 μL of pre-treated Coagtrol N in a UV–visible cuvette. Each mixture was incubated at 37 °C in a water bath for 2 min, followed by the addition of 25 mM CaCl2 (50 μL). The clotting time for each sample was then determined.
The coagulation tests using Actin-FSL and PS particles were conducted throughout the year, specifically in February, May, July, and October of 2020. This approach aimed to demonstrate the fact that PS particles produce stable results across all seasons and environments (Figure 7).
For the blank control group, the procedure was slightly modified: 50 μL of Coagtrol N was pre-warmed in a UV–visible cuvette at 37 °C for 1 min before adding 50 μL of the ellagic acid-based HEPES buffer solution. After a further incubation of 2 min at 37 °C, CaCl2 (25 mM, 50 μL) was added, and the clotting time was calculated based on the change in absorbance at 500 nm. The coagulation time was defined from the moment CaCl2 was added until the absorbance reached half of the maximum difference observed after reagent addition. The normal reference ranges for the APTT were confirmed according to the Actin FSL reagent kit instructions, serving as a positive control.
All statistical analyses were carried out using Tukey methods.
2.6. Preparation of Blood Clots Specimen
To investigate the morphological differences in the fibrin network and fiber structure following the addition of Actin FSL and PS particle solutions, scanning electron microscopy (SEM) was employed for high-resolution visualization. For both the Actin FSL and PS particle groups, fibrin clots were carefully transferred from the UV–visible cuvette to polystyrene cell culture dishes for fixation. The fixation process involved immersing the specimens in phosphate-buffered saline (PBS) containing 2% glutaraldehyde for 60 min at room temperature. Following fixation, the samples were washed briefly in PBS twice for 5 min each, and then rinsed in distilled water twice for 5 min each. The specimens underwent a series of dehydration steps in ethanol solutions of increasing concentrations (70%, 95%, and 100%), spending 10 min in each solution, followed by a 10-min immersion in pure acetone. After dehydration, the specimens were air-dried for 60 min at room temperature. The dried specimens were then mounted on aluminum stubs and sputter-coated with gold/palladium for 1 min (SC-701 MKII, Tokyo, Japan) to prepare them for SEM observation. The acceleration voltage was 20 kV (Figure 3d–f).
4. Discussion
The surface of PS particles prepared at the highest concentration, as seen in SEM images (specifically,
Figure 3f), showed wrinkles. This characteristic is considered to be a result of the preparation method rather than an intrinsic property of the particles. The smallest particles, measuring 906 nm in diameter and produced at the lowest concentration of 2 mg/mL, exhibited the most negative zeta potential. Considering that particle size and zeta potential do not have a direct theoretical relationship, a change in surface structure could imply that these particles expose a greater number of PS moieties. A significant difference in coagulation times was observed between particles produced at 8 mg/mL and those at 2 mg/mL, as determined by Tukey’s method. The explanation for this finding is that smaller particles have a larger surface area relative to their volume, likely allowing for more PS moieties to be exposed. Given that PS acts as a trigger in coagulation, this finding is noteworthy. From these observations, it can be concluded that PS particles prepared at a concentration of 2 mg/mL are notably effective. Their smaller size and larger specific surface area mean they expose more PS moieties per unit area than those prepared at higher concentrations. This efficiency in exposing PS moieties suggests they could be more effective in applications where coagulation time is a critical factor.
From the perspective of coagulation time, a concentration of 0.06 mg/mL exhibited the shortest coagulation time. PS groups act as an accelerator in blood coagulation, suggesting a negative correlation between PS particle concentration and coagulation time up to a certain concentration [
19]. However, since PS groups strongly trap calcium ions, an essential factor in coagulation, exceeding a certain concentration can deplete calcium ions in the system [
20,
21]. This depletion would predictably shift the correlation from negative to positive beyond a certain PS particle concentration. In this experiment, the optimal balance was found at a concentration of 0.06 mg/mL. It is important to note that this balance can vary with the particle size and the amount of PS moieties introduced, requiring evaluation with each new batch synthesis.
In the examination of the morphology of fibrin clots, no significant differences were observed between the samples created using Actin-FSL and those with PS particles. The only distinction was the presence of PS particles. In both types of samples, fibers forming a uniform network were identified, suggesting that PS particles may induce coagulation through a mechanism similar to that of coagulation reagents [
22,
23]. This observation implies that PS particles could be functioning in a manner akin to traditional coagulation factors, facilitating the formation of uniformly structured fibrin clots.
To evaluate the impact of external environmental conditions on the coagulation time of PS particles, the same coagulation test was conducted at four different time points over a year, and no differences were observed. All tests were carried out in Tsukuba City, Japan, in February, May, July, and October. The average temperature, average highest temperature, average lowest temperature, and average humidity for each month in 2020 are summarized in
Table 1.
The evaluation conducted at different time points showed no significant difference in coagulation time for Actin-FSL, indicating stable performance across the seasons. Similarly, PS particles exhibited no significant differences at all time points. Although all measurements were conducted in a laboratory where temperature and humidity were controlled to be constant, it was anticipated that there could be slight variations due to the actual seasonal temperatures and humidity levels. Thus, the experiment was not designed to expose the systems to seasonal external environments directly but rather aimed to discuss the potential impact of seasonal variations at the laboratory level on the evaluation. Moreover, the PS particles used in this test were from the same batch and stored in a lyophilized state, indicating that PS particles maintain their activity for at least one year without significant loss of function.
Aizhen Yang et al. successfully demonstrated that apoptotic cells had the ability to accelerate blood coagulation [
25]. Aizhen Yang et al. revealed that Factor XII (FXII) preferentially binds to apoptotic cells and is rapidly activated, demonstrating the contribution of apoptotic cells to coagulation enhancement. While thrombin generation is initiated by two pathways associated with vascular damage and blood-derived factors, this research indicates that phosphatidylserine (PS) in apoptotic cells functions as a novel activator of FXII. Furthermore, it was shown that FXII plays a crucial role in coagulation mediated by apoptotic cells. Considering the importance of the rapid clearance of apoptotic cells in maintaining anti-inflammatory and antithrombotic states, especially in the context of increased apoptotic cells due to autoimmune diseases, chemotherapy, and inflammation, this could lead to the development of new therapeutic strategies [
26,
27]. The findings suggest that PS particles, which mimic apoptotic cells as reported in this study, not only promote blood coagulation through a similar mechanism but also have potential applications in the body since they are cleared from tissues by immune cells with PS receptors, extending beyond coagulants. Such potential of PS can be combined with other therapeutic strategies and pave the way to developing new medicines and medical devices [
28,
29].











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}