
Enantioselective Biomimetic Structures Inspired by Oxi-Dase-Type Metalloenzymes, Utilizing Polynuclear Compounds Containing Copper (II) and Manganese (II) Ions as Building Blocks

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Biomimetic Compounds
2.3. Physical Measurements
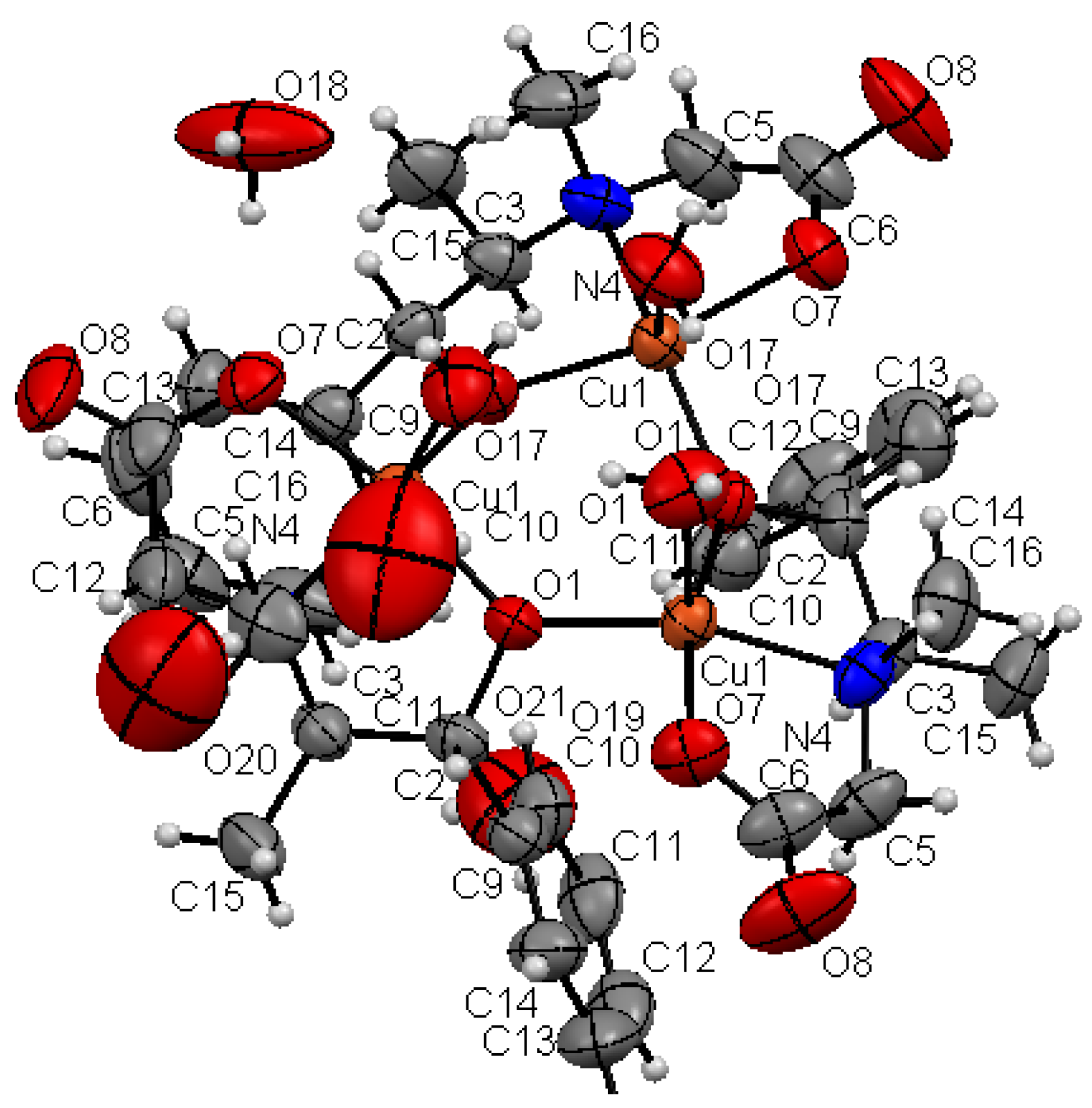
2.4. X-ray Crystallographic Study
2.5. Electrochemical Analyses
2.6. Catalytic Activities of the Biomimetic Models
3. Results
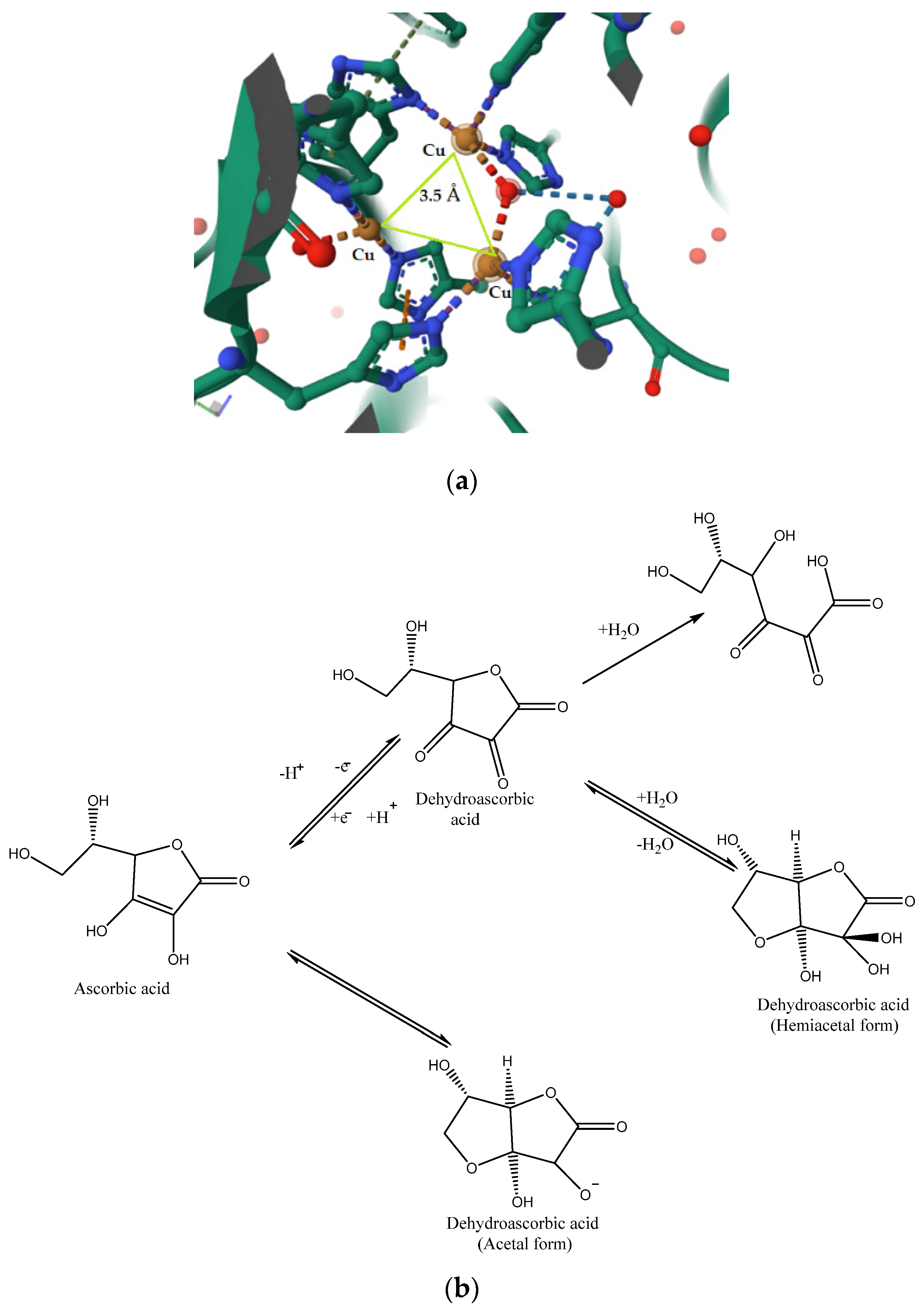
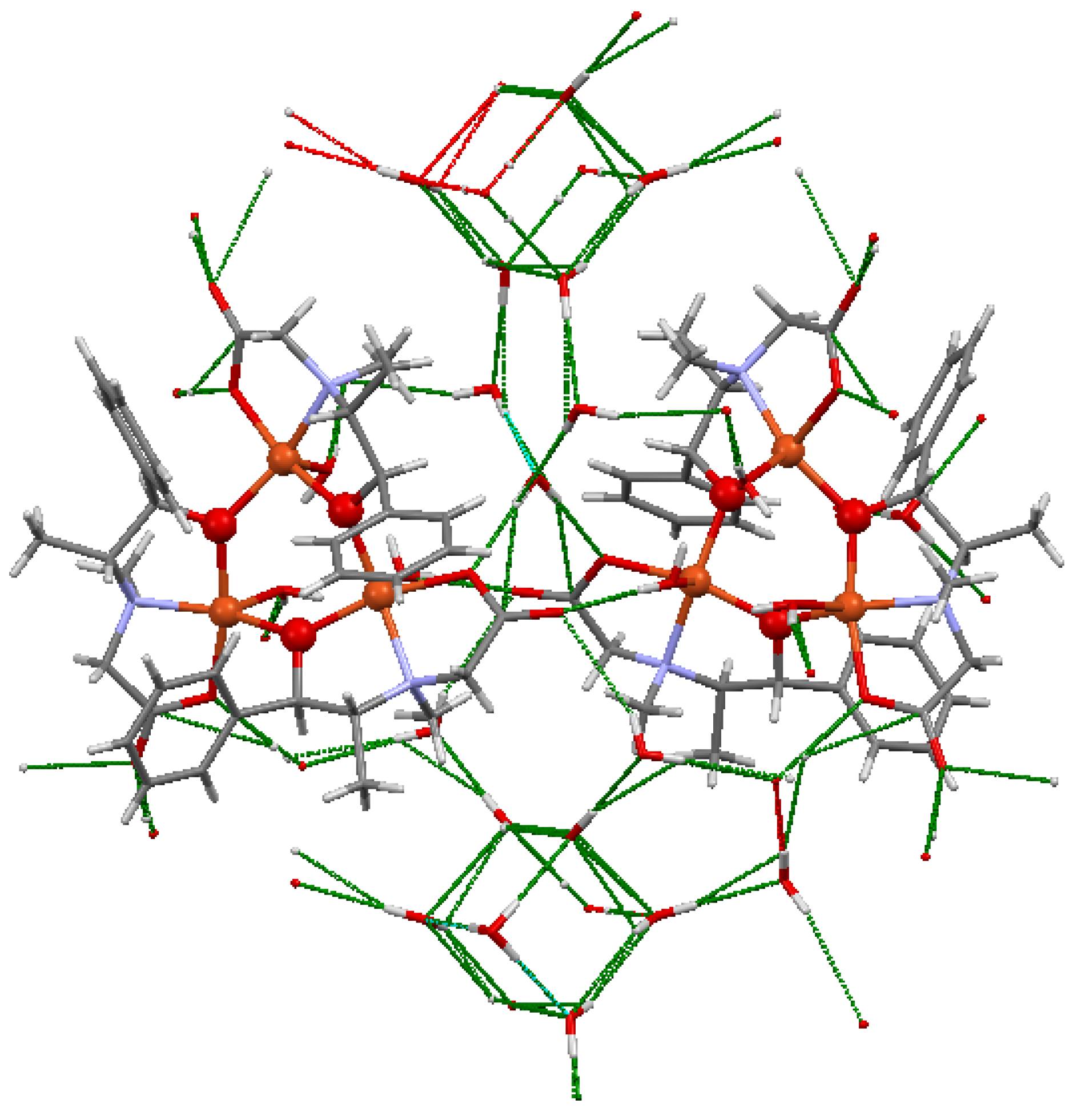
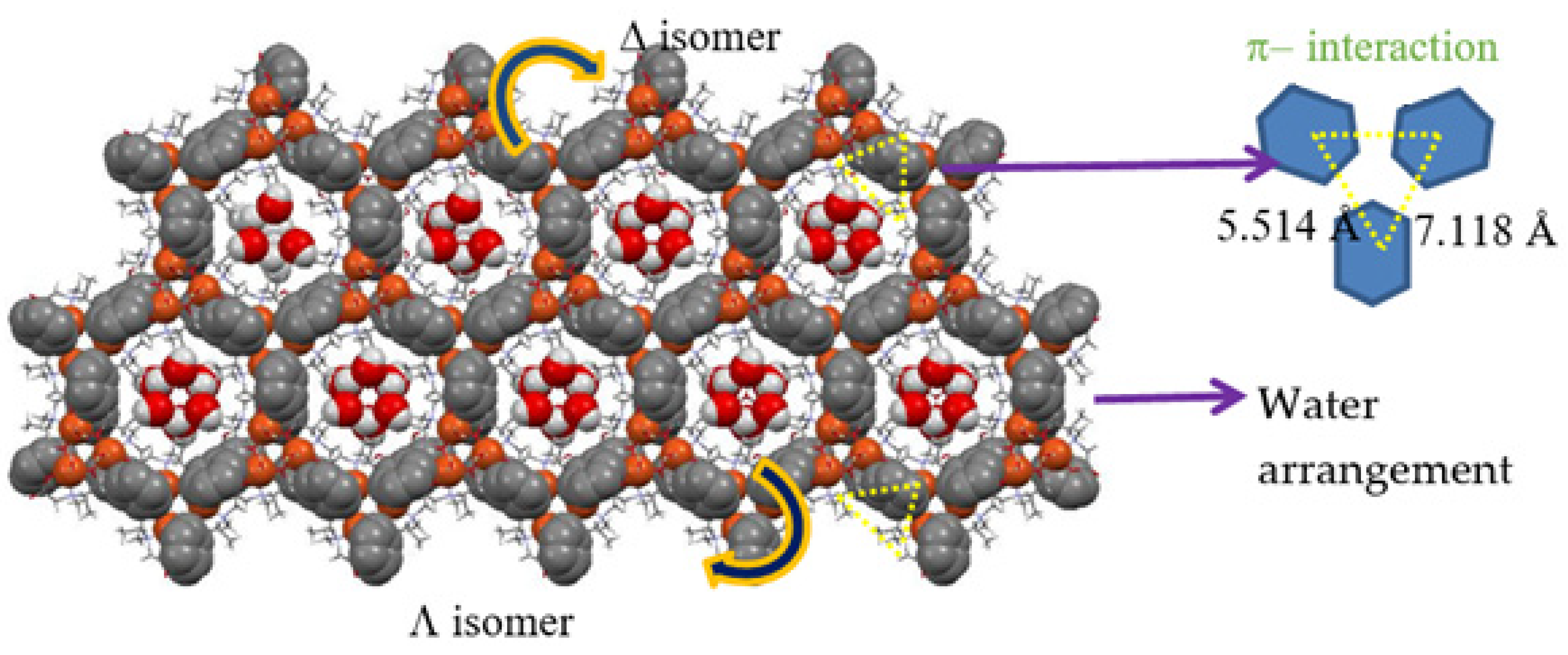
3.1. Crystal Structure of the AO Biomimetic Model
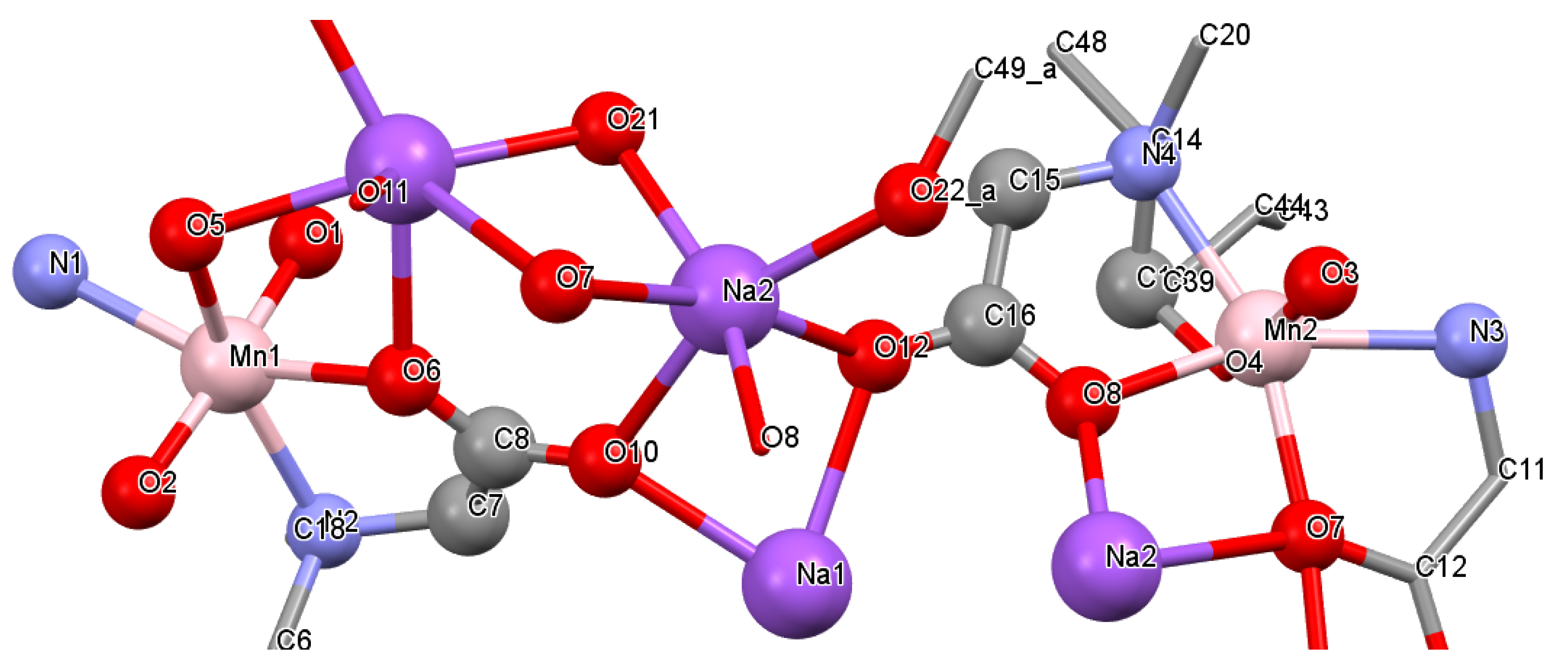
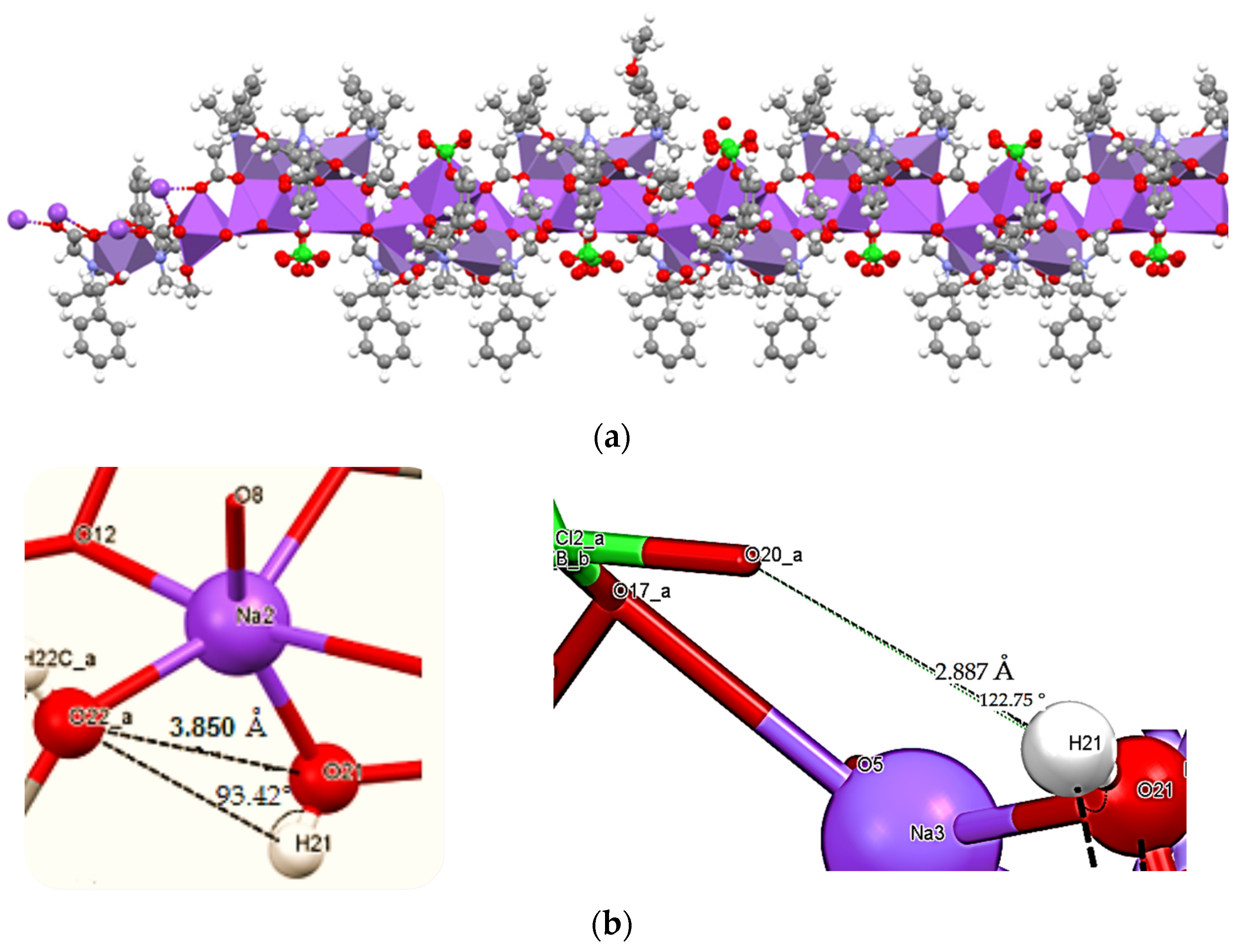
3.2. Crystal Structure of the Catalase Biomimetic Model and Spectroscopic Characterization for Its Enaniomeric Corresponding Compound
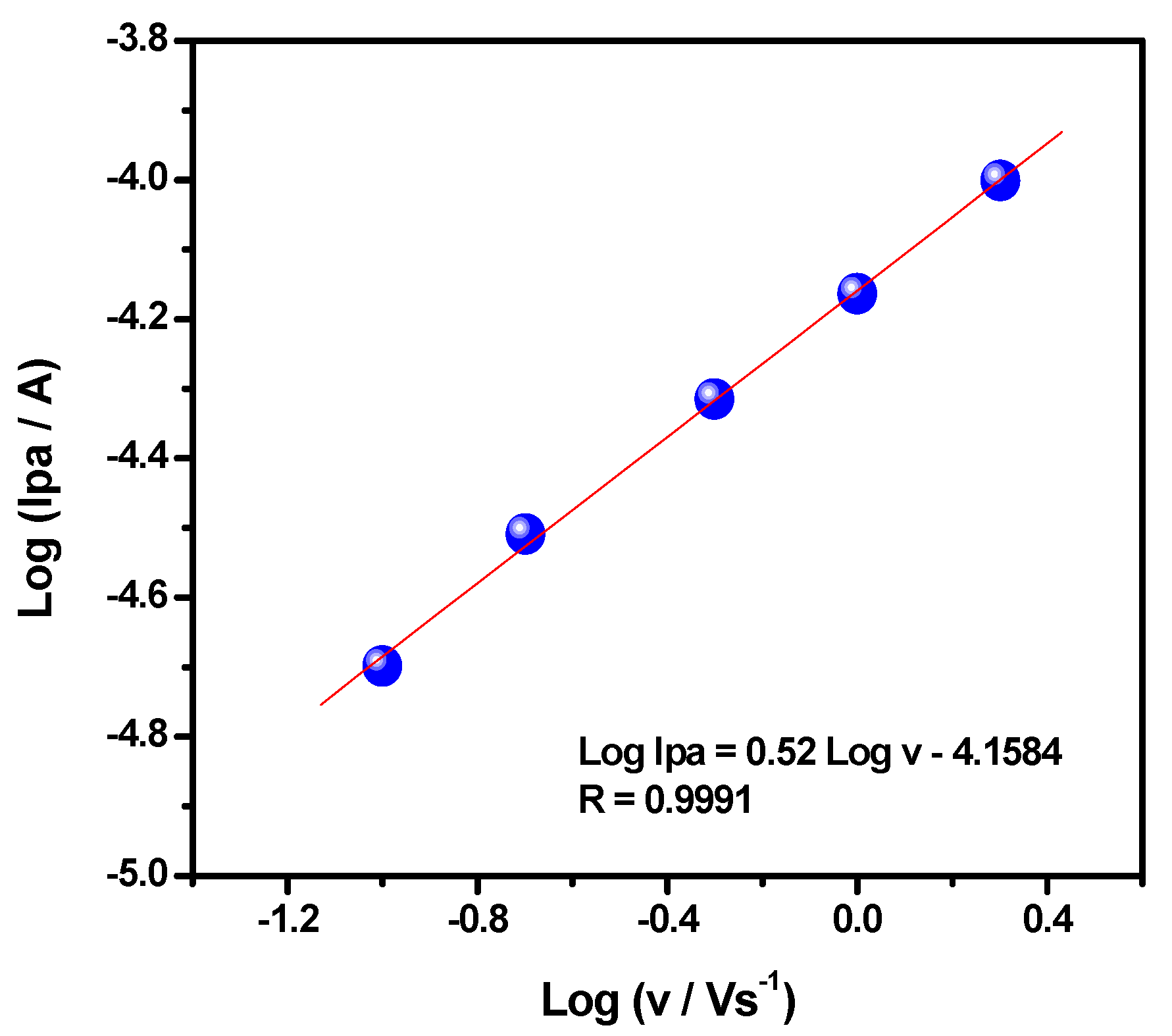
3.3. Electrochemical Studies of the AO Biomimetic Model
3.4. Electrochemical Studies of the Catalase Biomimetic Model
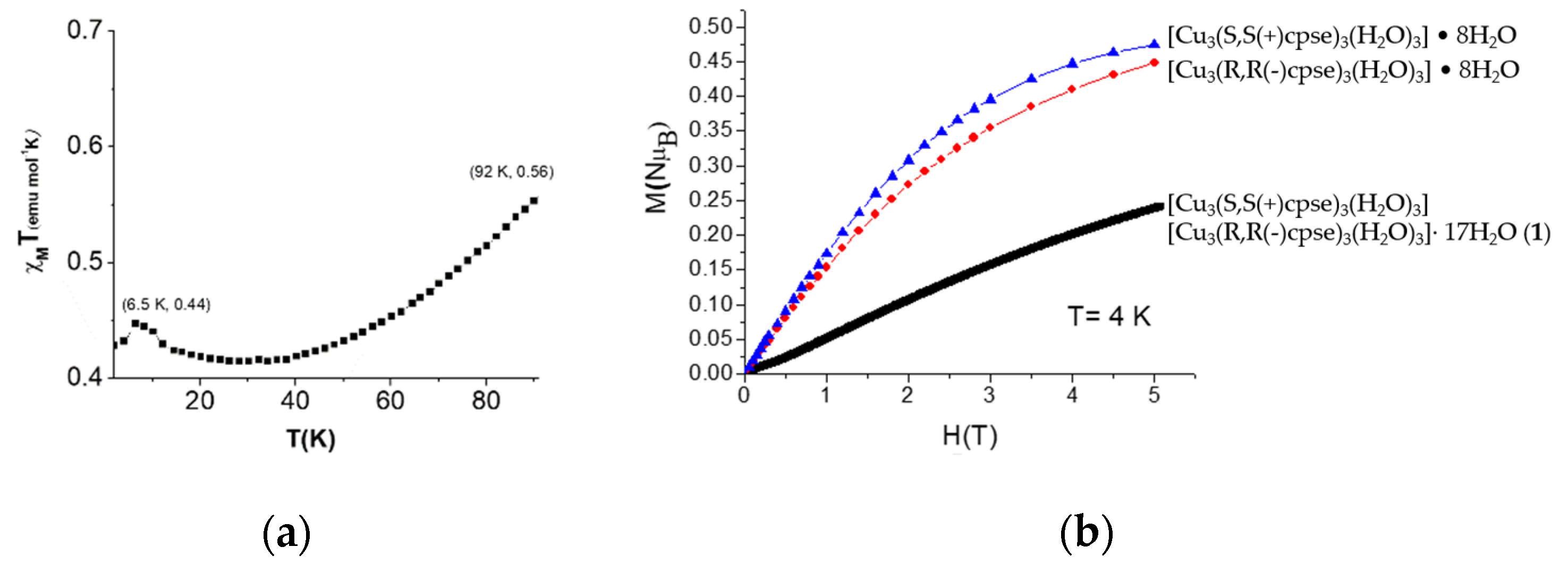
3.5. Magnetic Properties of the AO Biomimetic Model
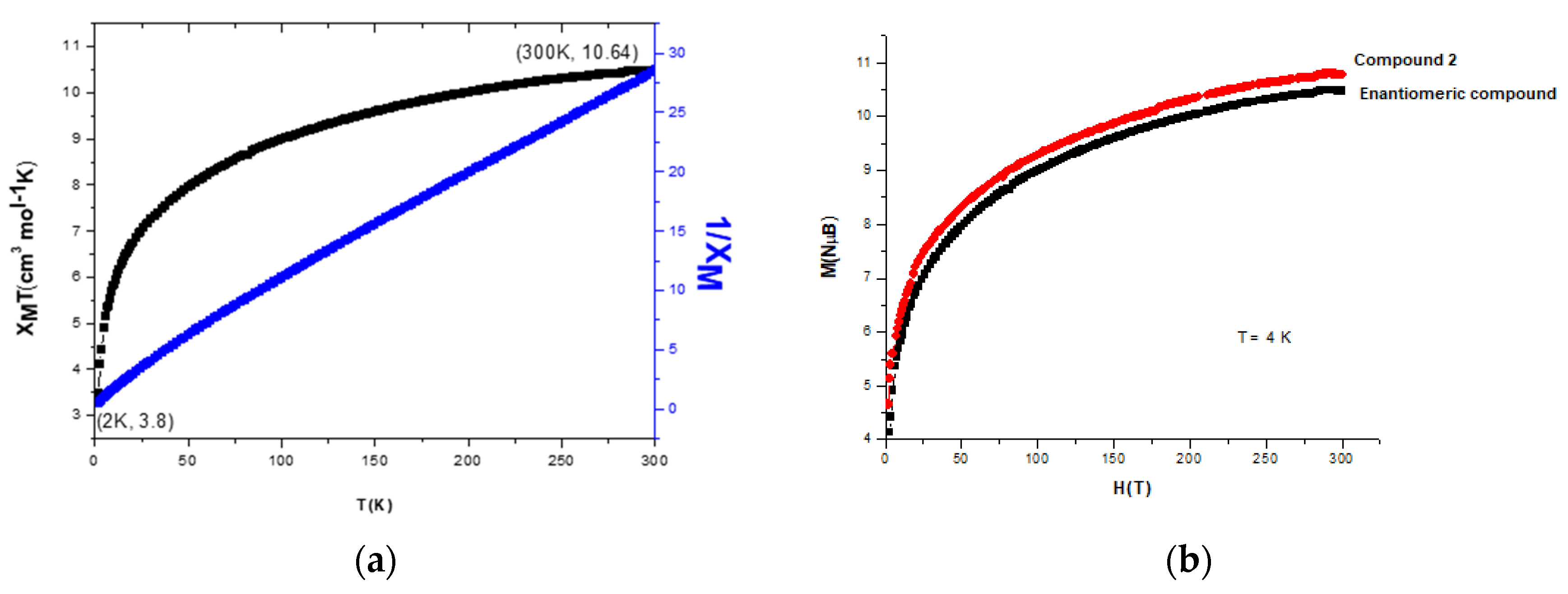
3.6. Magnetic Properties of the Catalase Biomimetic Model
3.7. Assessment of the Activity of the AO Biomimetic Model
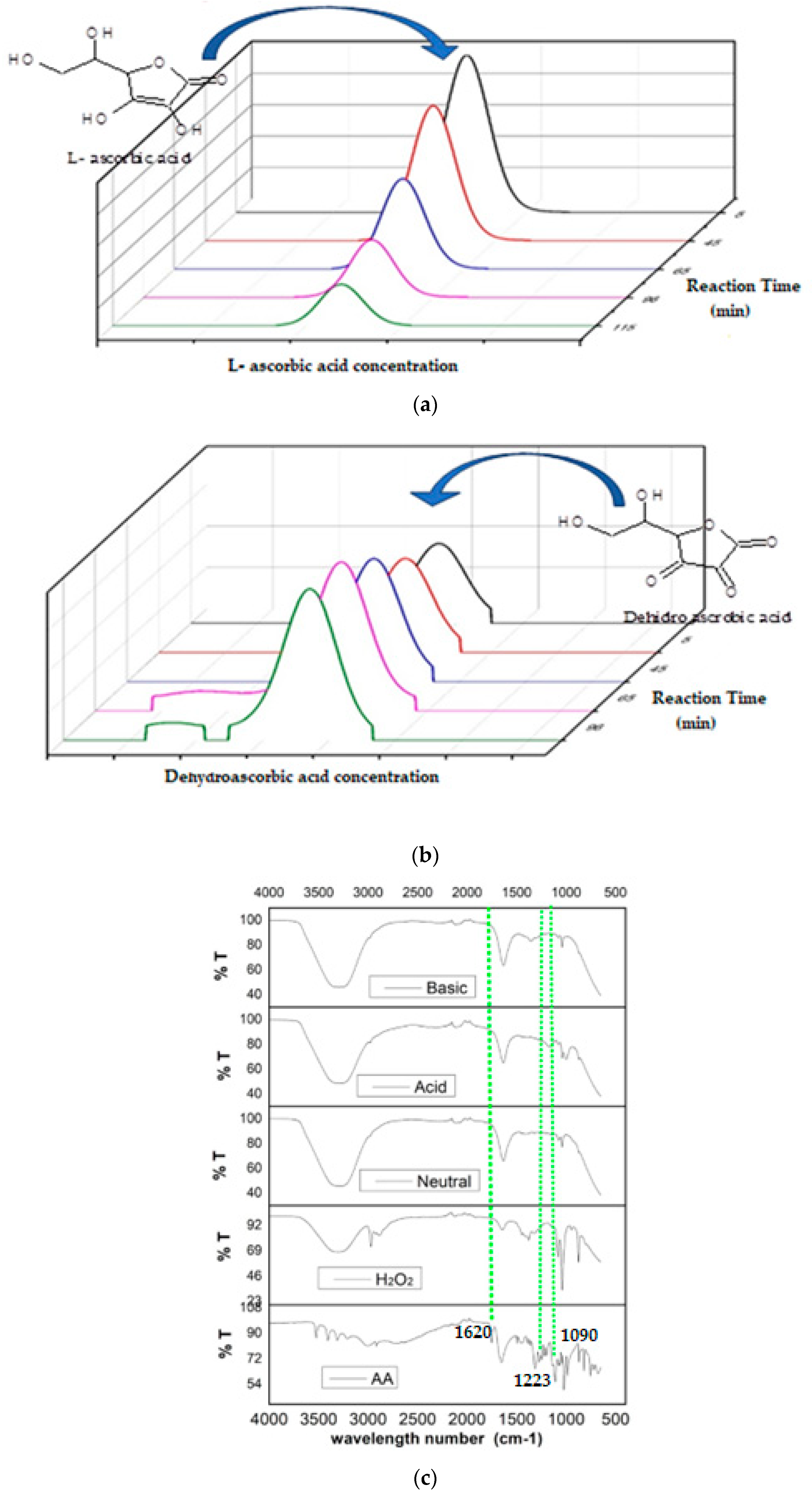
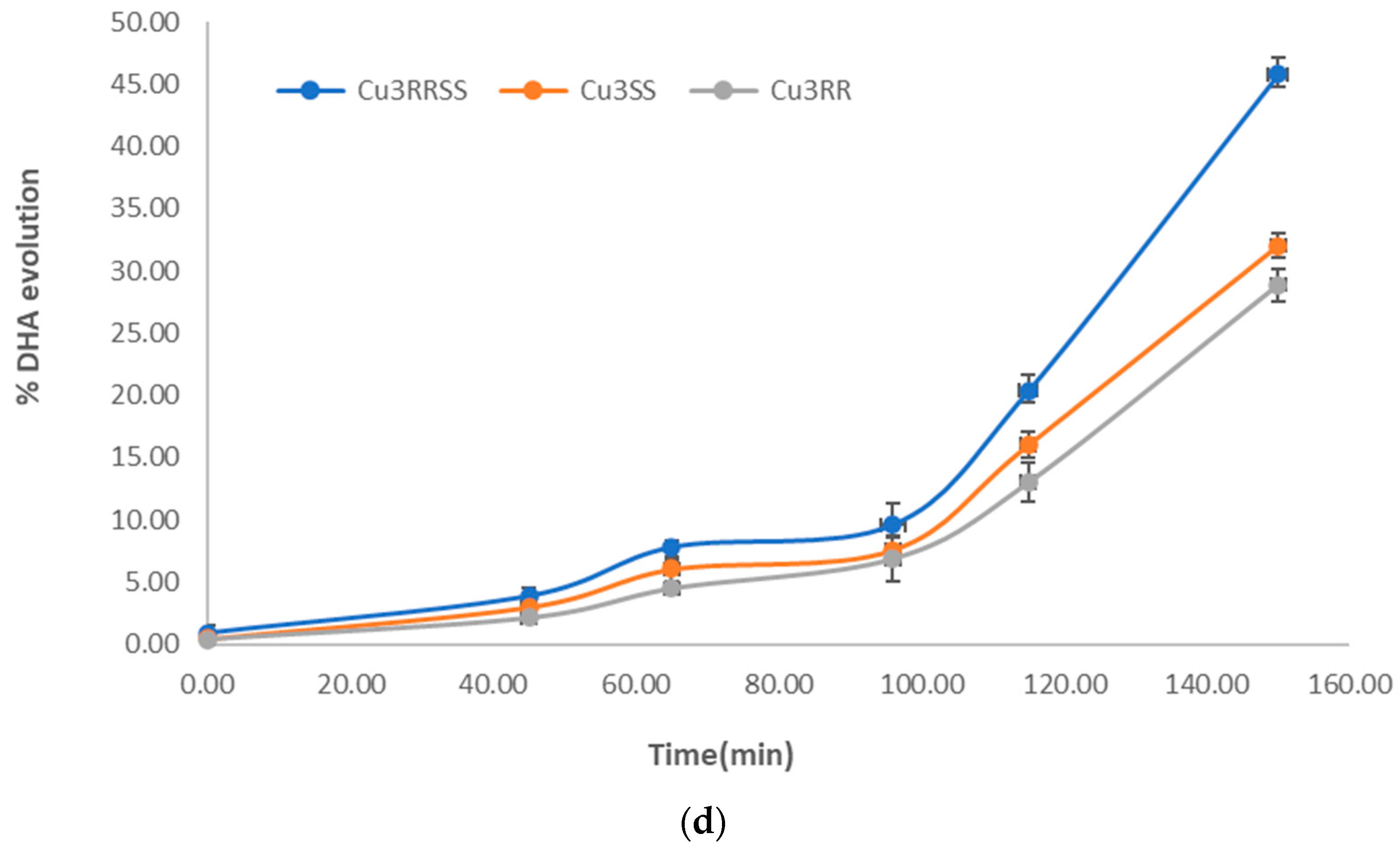
3.8. Assessment of the Activity of the Catalase Biomimetic Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Benkovic, S.J.; Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-M.; Han, S.-S.; Kim, H.-S. Industrial applications of enzyme biocatalysis: Current status and future aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Franssen, M.C.R.; Steunenberg, P.; Scott, E.L.; Zuilhof, H.; Sanders, J.P.M. Immobilised enzymes in biorenewables production. Chem. Soc. Rev. 2013, 42, 6491. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, Y.; Xu, Z.-J.; Che, C.-M. Iron(III)–salan complexes catalysed highly enantioselective fluorination and hydroxylation of β-keto esters and N-Boc oxindoles. Chem. Commun. 2014, 50, 7870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187. [Google Scholar] [CrossRef] [PubMed]
- Balcells, D. Insight into metal-catalyzed water oxidation from a DFT perspective. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 115–173. [Google Scholar] [CrossRef]
- Phale, P.S.; Sharma, A.; Gautam, K. Microbial degradation of xenobiotics like aromatic pollutants from the terrestrial environments. In Pharmaceuticals and Personal Care Products: Waste Management and Treatment Technology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 259–278. [Google Scholar] [CrossRef]
- Marqués, J.; Cortés, A.; Pejenaute, Á.; Zalba, G. Implications of NADPH oxidase 5 in vascular diseases. Int. J. Biochem. Cell Biol. 2020, 128, 105851. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Couto, S.; Toca Herrera, J.L. Industrial and biotechnological applications of laccases: A review. Biotechnol. Adv. 2006, 24, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Kunamneni, A.; Plou, F.; Ballesteros, A.; Alcalde, M. Laccases and Their Applications: A Patent Review. Recent Pat. Biotechnol. 2008, 2, 10–24. [Google Scholar] [CrossRef]
- Messerschmidt, A.; Ladenstein, R.; Huber, R.; Bolognesi, M.; Avigliano, L.; Petruzzelli, R.; Rossi, A.; Finazzi-Agró, A. Refined crystal structure of ascorbate oxidase at 1.9 Å resolution. J. Mol. Biol. 1992, 224, 179–205. [Google Scholar] [CrossRef]
- Davey, M.W.; Montagu, M.; Van Inz, D.; Sanmartin, M.; Kanellis, A.; Smirnoff, N.; Benzie, I.J.; Strain, J.J.; Favell, D.; Fletcher, J. PlantL-ascorbic acid: Chemistry, function, metabolism, bioavailability and effects of processing. J. Sci. Food Agric. 2000, 80, 825–860. [Google Scholar] [CrossRef]
- Lloyd, D.; Vainikka, T.; Murtomäki, L.; Kontturi, K.; Ahlberg, E. The kinetics of the Cu2+/Cu+ redox couple in deep eutectic solvents. Electrochim. Acta 2011, 56, 4942–4948. [Google Scholar] [CrossRef]
- Galicia, M.; González-Fuentes, M.A.; Valencia, D.P.; González, F.J. The effect of substituents on the anodic oxidation of aliphatic carboxylates and the passage towards a pseudo-Kolbe reaction. J. Electroanal. Chem. 2012, 672, 28–33. [Google Scholar] [CrossRef]
- Conesa, A.; Punt, P.J.; van den Hondel, C.A.M.J.J. Fungal peroxidases: Molecular aspects and applications. J. Biotechnol. 2002, 93, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, M. Review: Lignin conversion by manganese peroxidase (MnP). Enzym. Microb. Technol. 2002, 30, 454–466. [Google Scholar] [CrossRef]
- Wariishi, H.; Valli, K.; Gold, M.H. In vitro depolymerization of lignin by manganese peroxidase of Phanerochaete chrysosporium. Biochem. Biophys. Res. Commun. 1991, 176, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Torres, Y.; Acosta, J.; Huerta, L.; Toscano, A.; González, F.J.; Behrens, N.B. Experimental data on synthesis and characterization of chiral dinuclear manganese (II-II) compounds as biomimetic models of the active center of catalase. Data Brief 2020, 28, 104883. [Google Scholar] [CrossRef] [PubMed]
- Morgan Chan, Z.; Kitchaev, D.A.; Nelson Weker, J.; Schnedermann, C.; Lim, K.; Ceder, G.; Tumas, W.; Toney, M.F.; Nocera, D.G. Electrochemical trapping of metastable Mn3+ ions for activation of MnO2 oxygen evolution catalysts. Proc. Natl. Acad. Sci. USA 2018, 115, E5261–E5268. [Google Scholar] [CrossRef] [PubMed]
- Valencia, I.; Ávila-Torres, Y.; Barba-Behrens, N.; Garzón, I.L. Structural, vibrational, and electronic properties of an uncoordinated pseudoephedrine derivative and its mononuclear and trinuclear copper(II)-coordinated compounds: A combined theoretical and experimental study. J. Mol. Struct. 2014, 1076, 387–395. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Panayotov, D.A.; Mihaylov, M.Y.; Ivanova, E.Z.; Chakarova, K.K.; Andonova, S.M.; Drenchev, N.L. Power of Infrared and Raman Spectroscopies to Characterize Metal-Organic Frameworks and Investigate Their Interaction with Guest Molecules. Chem. Rev. 2021, 121, 1286–1424. [Google Scholar] [CrossRef]
- Ávila-Torres, Y.; López-Sandoval, H.; Mijangos, E.; Quintanar, L.; Rodríguez, E.E.; Flores-Parra, A.; Contreras, R.; Vicente, R.; Rikken, G.L.; Barba-Behrens, N. Structure and magnetic properties of copper(II) and cobalt(II) coordination compounds derived from optically active tridentate ligands. Polyhedron 2013, 51, 298–306. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Herr, P.; Kerzig, C.; Larsen, C.B.; Häussinger, D.; Wenger, O.S. Manganese(I) complexes with metal-to-ligand charge transfer luminescence and photoreactivity. Nat. Chem. 2021, 13, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizzi, L. The ferrocenium/ferrocene couple: A versatile redox switch. ChemTexts 2020, 6, 22. [Google Scholar] [CrossRef]
- Julve, M.; Gleizes, A.; Chamoreau, L.M.; Ruiz, E.; Verdaguer, M. Antiferromagnetic Interactions in Copper(II) µ-Oxalato Dinuclear Complexes: The Role of the Counterion. Eur. J. Inorg. Chem. 2018, 2018, 509–516. [Google Scholar] [CrossRef]
- Zhao, X.G.; Richardson, W.H.; Chen, J.-L.; Li, J.; Noodleman, L.; Tsai, H.-L.; Hendrickson, D.N. Density Functional Calculations of Electronic Structure, Charge Distribution and Spin Coupling in Manganese−Oxo Dimer Complexes. Inorg. Chem. 1997, 36, 1198–1217. [Google Scholar] [CrossRef] [PubMed]
- Ramidi, P.; Felton, C.M.; Subedi, B.P.; Zhou, H.; Tian, Z.R.; Gartia, Y.; Pierce, B.S.; Ghosh, A. Synthesis and characterization of manganese(III) and high-valent manganese-oxo complexes and their roles in conversion of alkenes to cyclic carbonates. J. CO2 Util. 2015, 9, 48–57. [Google Scholar] [CrossRef]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, A.E.-M.M.; Shaban, S.Y.; Ibrahim, M.M. Synthesis, characterization, and ascorbic acid oxidase biomimetic catalytic activity of cobalt(III) oxime complexes. J. Coord. Chem. 2011, 64, 3376–3392. [Google Scholar] [CrossRef]
- Robert, A.; Loock, B.; Momenteau, M.; Meunier, B. Catalase modeling with metalloporphyrin complexes having an oxygen ligand in a proximal position. Comparison with complexes containing a proximal nitrogen. Inorg. Chem. 1991, 30, 706–711. [Google Scholar] [CrossRef]
- Balasubramanian, P.N.; Schmidt, E.S.; Bruice, T.C. Catalase modeling. 2. Dynamics of reaction of a water-soluble and non.mu.-oxo dimer forming manganese(III) porphyrin with hydrogen peroxide. J. Am. Chem. Soc. 1987, 109, 7865–7873. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | 2(C36H51N3O12Cu3) 17(H2O) (1) | C54.15 H87.30 Cl2Mn2N4Na3O26 (2) |
|---|---|---|
| Formula weight (g mol−1) | 2123.14 | 1460.13 |
| Crystal size (mm3) | 0.20 × 0.15 × 0.15 | |
| Crystal color | Blue | dark purple |
| Crystal system | Trigonal | Monoclinic |
| Space group | R-32 c | C2 |
| Unit cell dimensions | ||
| a (Å) | 15.5811 (2) | 21.5749 (8) |
| 15.5811 (2) | ||
| 72.7841 (9) | ||
| b (Å) | 15.5811 (2) | 18.8865 (5) |
| c (Å) | 72.7841 (9) | 17.6692 (6) |
| α (°) | 90.0 | 90.0 |
| β (°) | 90.0 | 113.624 (4) |
| γ (°) | 120.0 | 90.0 |
| V (Å3) | 15302.5 (2) | 6596.4 (4) |
| Z | 6 | 4 |
| Dcalc (g/cm3) | 1.382 | 1.409 |
| F (000) | 3897 | 3061.0 |
| Temp (K) | 293 (2) | 130 |
| θ range (°) | 1.87–27.489 | 3.3855–29.4984 |
| Index range | −20 ≤ h ≤ 20 | −27 ≤ h ≤ 24 |
| −21 ≤ k ≤ 21 | ||
| −50 ≤ l ≤ 50 | ||
| −20 ≤ k ≤ 20 | −25 ≤ h ≤ 24 | |
| −21 ≤ k ≤ 21 | ||
| −50 ≤ l ≤ 50 | ||
| −78 ≤ l ≤ 94 | −24 ≤ l ≤ 21 | |
| −21 ≤ k ≤ 21 | ||
| −50 ≤ l ≤ 50 | ||
| Reflections measured | 45418 | 27019 |
| Independent reflections | 3897 | 15140 |
| Reflections | 3146 | 15139 |
| Rint | 0.0679 | 0.0605 |
| R | 0.0466 | 0.0621 |
| Rw | 0.1346 | 0.1438 |
| S | 1.04 | 0.926 |
| ∆ρ Maximum (e/Å3) | 0.78 | ------ |
| ∆ρ Minimum (e/Å3) | −0.43 | ------ |
| Flack parameter | ------ | ------ |
| Atoms | Angles (°) |
|---|---|
| O5–Cu1–N1 | 82.4 (1) |
| N1–Cu1–O3 | 84.5 (1) |
| O4–Cu1–O4A | 166.7 (1) |
| O5–Cu1–O4 | 97.0 (1) |
| N1–Cu1–O4 | 106.1 (1) |
| Atoms | Distances (Å) |
| Cu1A-O7 | 1.985 |
| Cu1-O17 | 2.322 |
| Cu1-O1 | 1.932 |
| Cu1-N4 | 2.018 |
| Cu1B-O7 | 1.947 |
| D–H...A | d(D–H) | D(D...A) | <(DHA) |
|---|---|---|---|
| O17—H171···O8 iii | 0.83 | 2.732 (7) | 165 |
| O17—H172···O17 ii | 0.83 | 2.755 (7) | 155 |
| O21—H211···O18 i | 0.82 | 2.972 (7) | 175 |
| O20—H201···O21 | 0.82 | 2.821 (7) | 179 |
| O18—H181···O7 | 0.87 | 2.771 (7) | 173 |
| O19—H191···O20 iv | 0.84 | 2.769 (7) | 178 |
| O20—H202···O20 iii | 0.82 | 3.062 (7) | 139 |
| Compound | Epa (V) | Epc (V) | ΔE (V) | E1/2 (V) |
|---|---|---|---|---|
| (+)S,S-H2cpse | 0.926 | — | — | — |
| (-)R,R-H2cpse | 0.927 | — | — | — |
| [Cu3(S,S(+)-cpse)3]∙8.5H2O | 0.145 a 0.414 b | −1.399 a, −0.982 a, −0.430 b | 1.254 a, 0.837 a, 0.844 b | −0.627 a, −0.415 a, −0.008 b |
| [Cu3(R,R(-)-cpse)3]∙8.5H2O | 0.193 a | −1.321 a, −0.958 a, −0.445 b | 1.514 a, 1.151 a | −0.564 a, −0.383 a |
| Compound 1 | 0.152 a 0.410 b | −1.389 a, −0.872 a, −0.445 b | 1.237 a, 0.72 a, 0.855 b | −0.619 a, −0.36 a, −0.017 b |
| Compound | Epa (V) | Epc (V) | ΔE (V) | E1/2 (V) |
|---|---|---|---|---|
| Compound 2 | 0.61 a | 0.47 a, 0.06, −0.6 | 0.14 a, 0.55, 0.01 | 0.07 a, 0.28, 0.005 |
| [Mn2(R,R(−)Hcpse)4(NaClO4)2(NaOH)(MeOH)]n·(EtOH)2n(MeOH)nH2On | 0.59 a | 0.45 a, 0.09, −0.5 | 0.14 a, 0.5, 0.09 | 0.07 a, 0.25, 0.045 |
| Oxidase Models | Oxidase Catalytic Activity (%) | [Cat] (%) | Catalase Activity (%) | [Cat] (%) |
|---|---|---|---|---|
| [CoLCl2], L = oxime [30] | 40 | 25 | ---- | ---- |
| [L’CoLBr]ClO4, L = oxime, L’ = triphenylphosphine | 50 | 25 | ---- | ---- |
| [CoL(SCN)(Br)], L = oxime [30] | 43 | 25 | ||
| [Mn(TMP)OAc], L = meso-tetraphenyl-porphyrinato [31] | ----- | ----- | 97 | 40 |
| [L’’’’M1(II)M2(II) Complexes], L = Chloro, M = Mn, Fe [31,32] | ----- | ----- | 28 | ---- |
| [Cu3(S,S(+)cpse)3(H2O)3] [Cu3(R,R(−)cpse)3(H2O)3]·17H2O | 45.5 | 5 | ||
| [Cu3(S,S(+)cpse)3(H2O)3]·8.5H2O | 32 | 5 | ||
| [Cu3(R, R(+)cpse)3(H2O)3]·8.5H2O | 28 | 5 | ||
| [Mn2(S,S(+)Hcpse)4(NaClO4)2(NaOH)(MeOH)]n·[(EtOH)2]n·[(MeOH)]n[H2O]n | 64.7 | 25 | ||
| [Mn2(R,R(+)Hcpse)4(NaClO4)2(NaOH)(MeOH)]n·[(EtOH)2]n·[(MeOH)]n[H2O]n | 27 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, D.; Acosta, J.; López-Sandoval, H.; Torres-Palma, R.A.; Ávila-Torres, Y. Enantioselective Biomimetic Structures Inspired by Oxi-Dase-Type Metalloenzymes, Utilizing Polynuclear Compounds Containing Copper (II) and Manganese (II) Ions as Building Blocks. Biomimetics 2023, 8, 423. https://doi.org/10.3390/biomimetics8050423
Gómez D, Acosta J, López-Sandoval H, Torres-Palma RA, Ávila-Torres Y. Enantioselective Biomimetic Structures Inspired by Oxi-Dase-Type Metalloenzymes, Utilizing Polynuclear Compounds Containing Copper (II) and Manganese (II) Ions as Building Blocks. Biomimetics. 2023; 8(5):423. https://doi.org/10.3390/biomimetics8050423
Chicago/Turabian StyleGómez, Didier, Jorge Acosta, Horacio López-Sandoval, Ricardo A. Torres-Palma, and Yenny Ávila-Torres. 2023. "Enantioselective Biomimetic Structures Inspired by Oxi-Dase-Type Metalloenzymes, Utilizing Polynuclear Compounds Containing Copper (II) and Manganese (II) Ions as Building Blocks" Biomimetics 8, no. 5: 423. https://doi.org/10.3390/biomimetics8050423
APA StyleGómez, D., Acosta, J., López-Sandoval, H., Torres-Palma, R. A., & Ávila-Torres, Y. (2023). Enantioselective Biomimetic Structures Inspired by Oxi-Dase-Type Metalloenzymes, Utilizing Polynuclear Compounds Containing Copper (II) and Manganese (II) Ions as Building Blocks. Biomimetics, 8(5), 423. https://doi.org/10.3390/biomimetics8050423