Kinetic Study of CO2 Hydration by Small-Molecule Catalysts with A Second Coordination Sphere that Mimic the Effect of the Thr-199 Residue of Carbonic Anhydrase


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Consideration
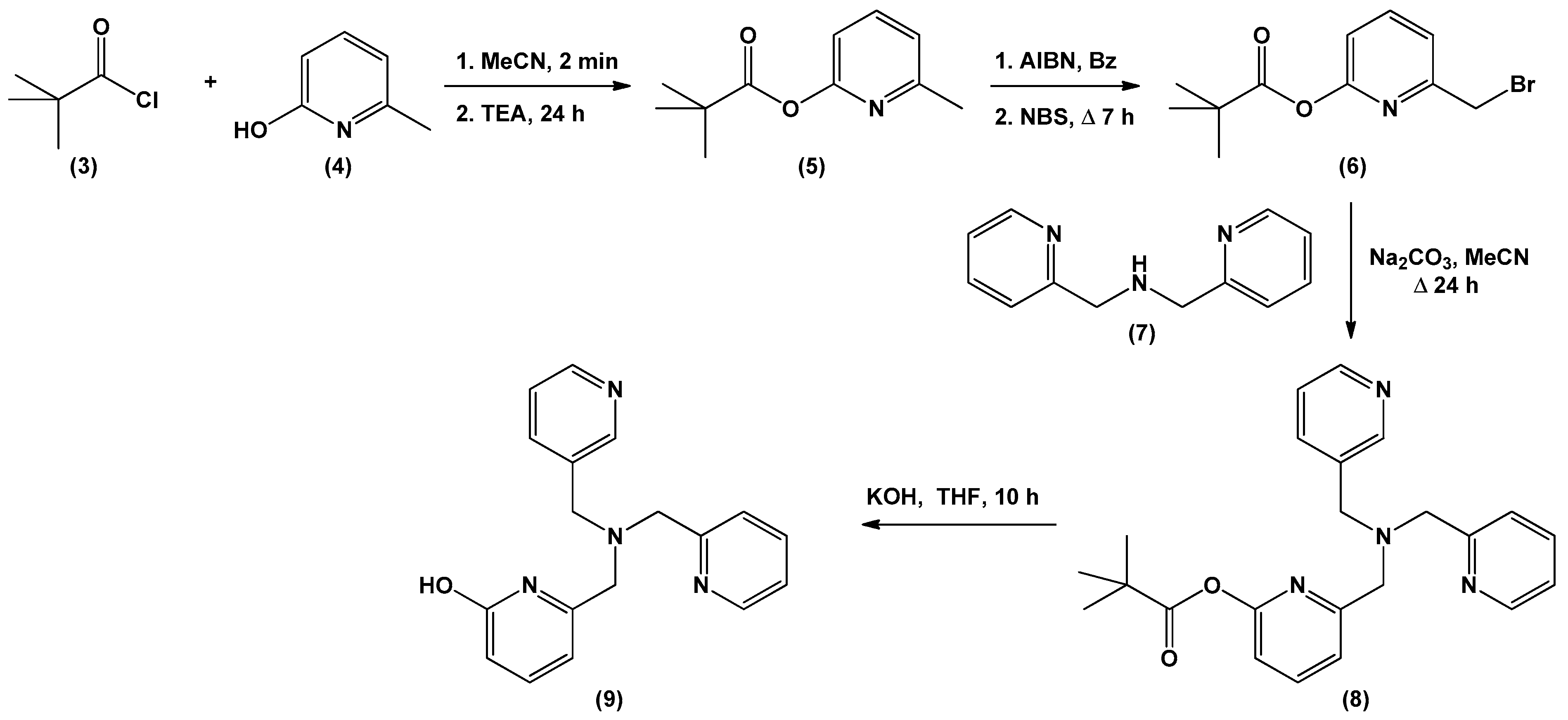
2.2. Materials Synthesis
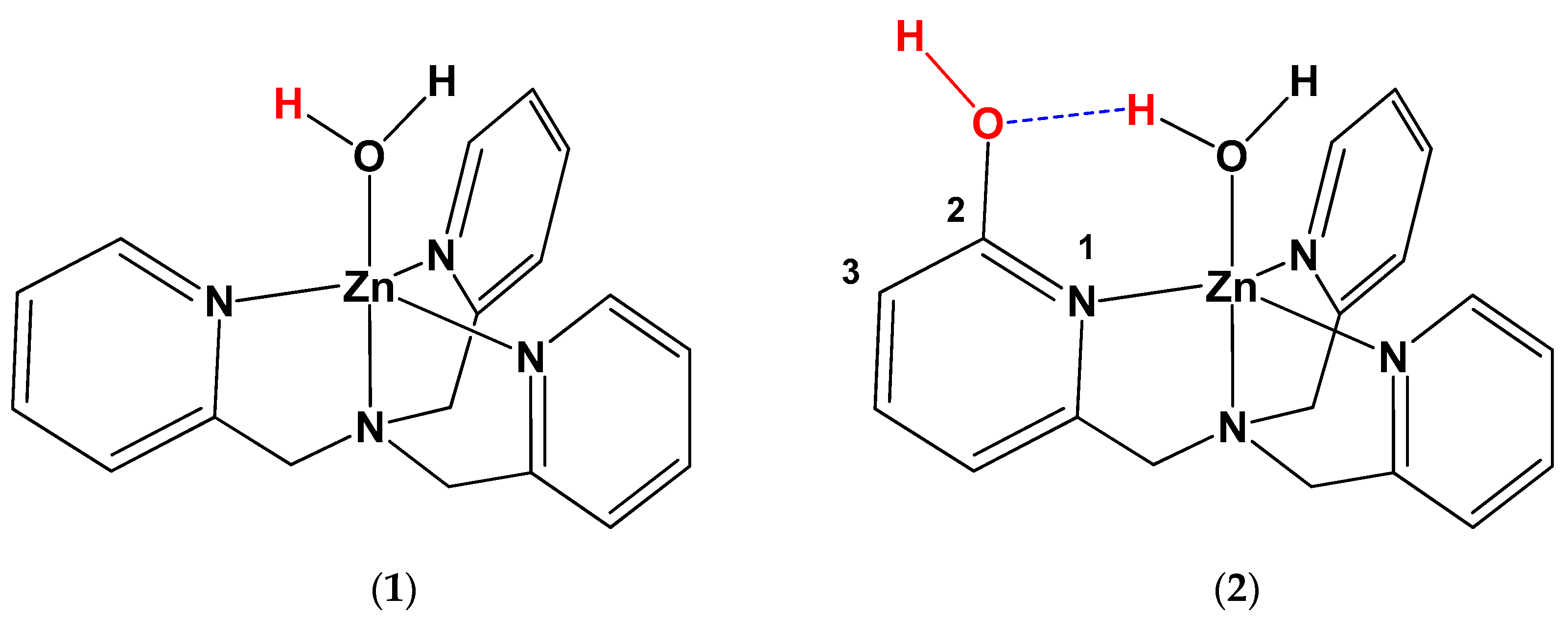
[(TPA)Zn(OH2)](ClO4)2 (1)
[(TPA-OH)Zn(OH2)](ClO4)2 (2)
2.3. Potentiometric pH Titration
2.4. Kinetic Measurements (Stopped-Flow Spectrophotometer)
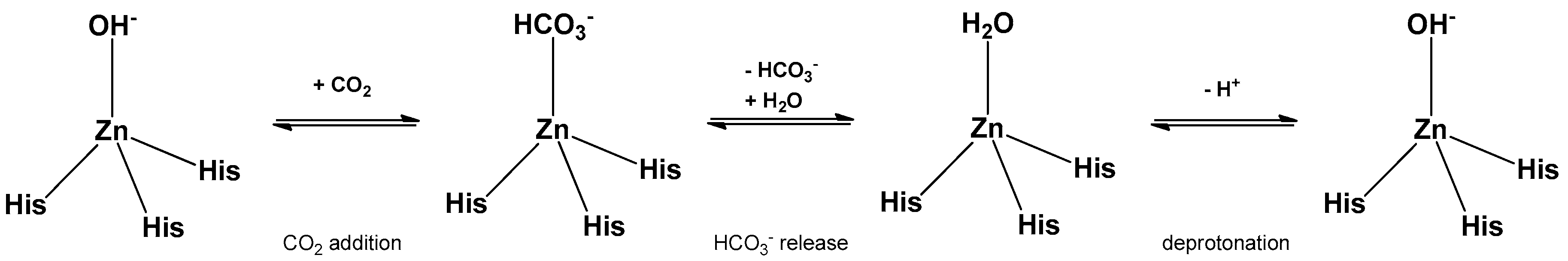
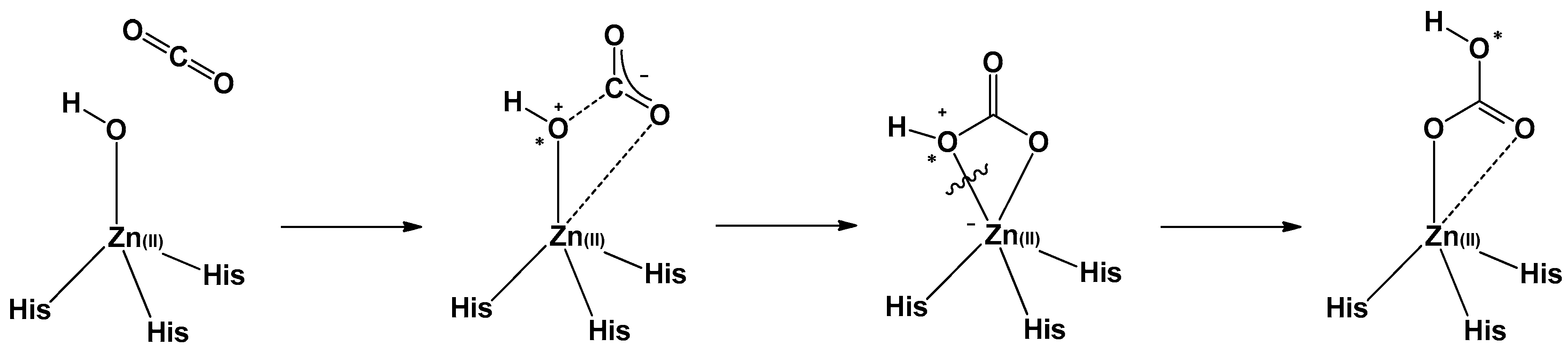
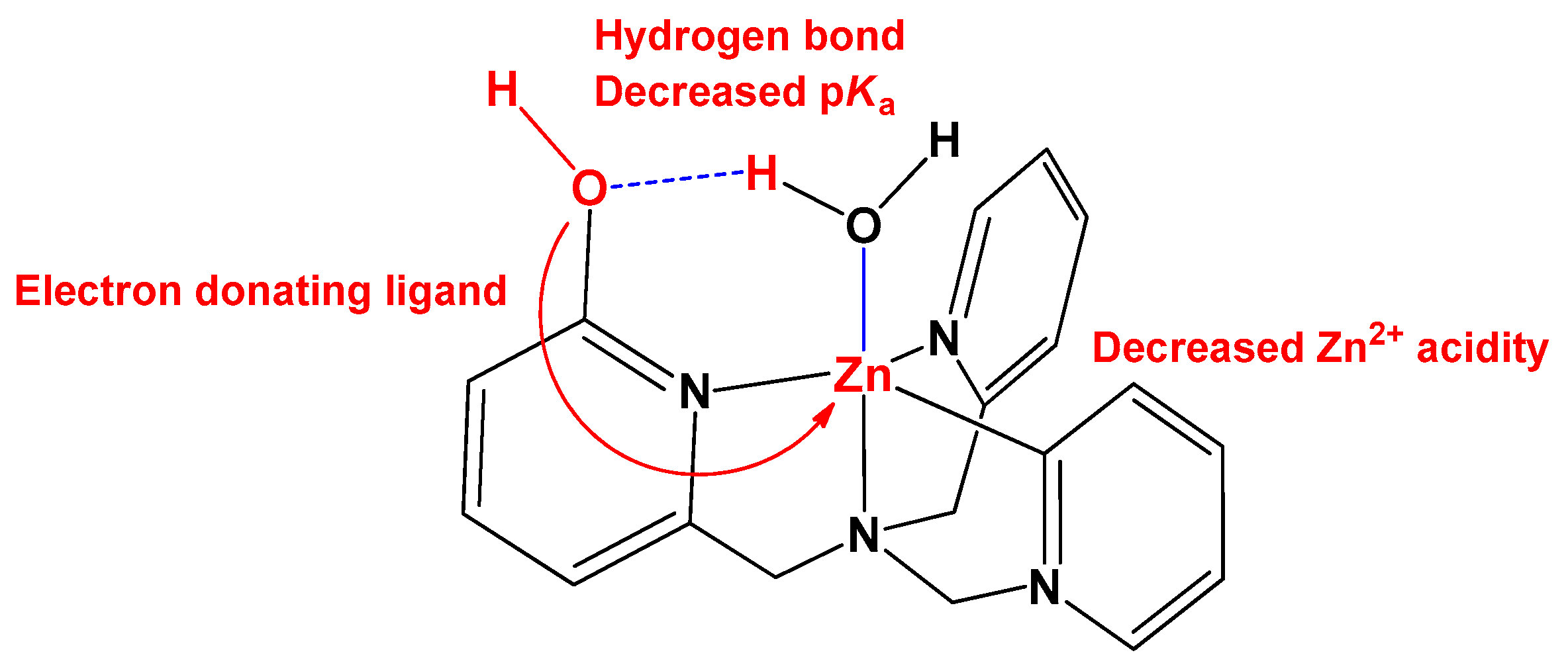
3. Results and Discussion
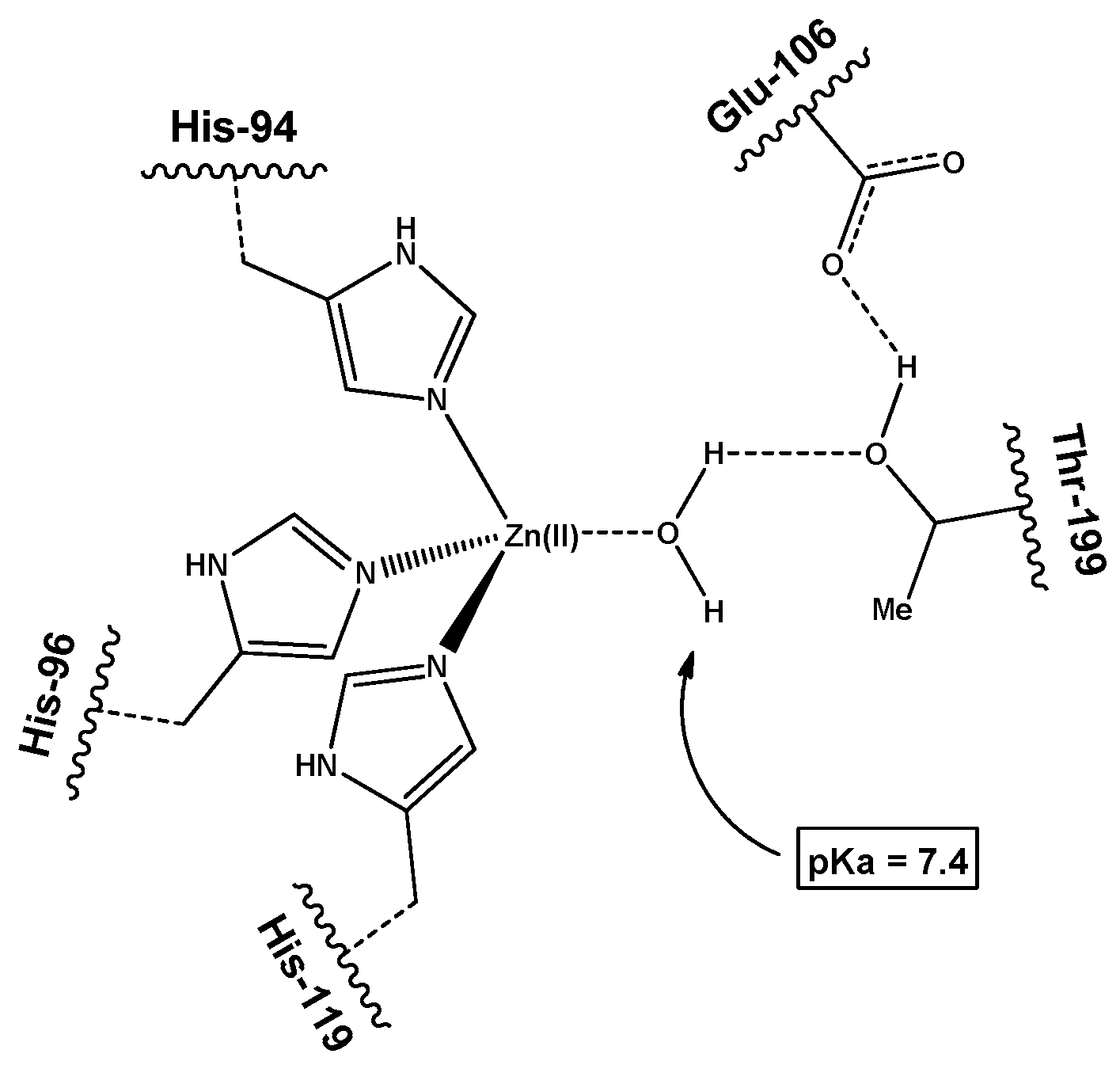
3.1. General Consideration
3.2. pKa Measurement
3.3. Kinetic Measurements
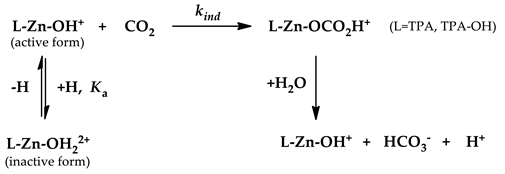
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A. Carbon capture and storage (CCS): The way forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, S.; Gao, H.; Yin, Q.; Liang, Z. A study of structure-activity relationships of aqueous diamine solutions with low heat of regeneration for post-combustion CO2 capture. Energy 2019, 167, 359–368. [Google Scholar] [CrossRef]
- Xiao, M.; Liu, H.; Gao, H.; Olson, W.; Liang, Z. CO2 capture with hybrid absorbents of low viscosity imidazolium-based ionic liquids and amine. Appl. Energy 2019, 235, 311–319. [Google Scholar] [CrossRef]
- Wang, J.; Sun, T.; Zhao, J.; Deng, S.; Li, K.; Xu, Y.; Fu, J. Thermodynamic considerations on MEA absorption: Whether thermodynamic cycle could be used as a tool for energy efficiency analysis. Energy 2019, 168, 380–392. [Google Scholar] [CrossRef]
- Oexmann, J.; Kather, A.; Linnenberg, S.; Liebenthal, U. Post-combustion CO2 capture: Chemical absorption processes in coal-fired steam power plants. Greenh. Gases Sci. Technol. 2012, 2, 80–98. [Google Scholar] [CrossRef]
- Wang, T.; Yu, W.; Liu, F.; Fang, M.; Farooq, M.; Luo, Z. Enhanced CO2 Absorption and Desorption by Monoethanolamine (MEA)-Based Nanoparticle Suspensions. Ind. Eng. Chem. Res. 2016, 55, 7830–7838. [Google Scholar] [CrossRef]
- Li, K.; Cousins, A.; Yu, H.; Feron, P.; Tade, M.; Luo, W.; Chen, J. Systematic study of aqueous monoethanolamine-based CO2 capture process: Model development and process improvement. Energy Sci. Eng. 2016, 4, 23–39. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, W.; Wang, K.; Wang, J. Studies of CO2 absorption/regeneration performances of novel aqueous monothanlamine (MEA)-based solutions. J. Clean. Prod. 2016, 112, 4012–4021. [Google Scholar] [CrossRef]
- Gupta, M.; da Silva, E.F.; Hartono, A.; Svendsen, H.F. Theoretical Study of Differential Enthalpy of Absorption of CO2 with MEA and MDEA as a Function of Temperature. J. Phys. Chem. B 2013, 117, 9457–9468. [Google Scholar] [CrossRef]
- Wang, M.; Wang, M.; Rao, N.; Li, J.; Li, J. Enhancement of CO2 capture performance of aqueous MEA by mixing with [NH2e-mim][BF4]. RSC Adv. 2018, 8, 1987–1992. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Stevens, G.W.; Caruso, F.; Kentish, S.E. The use of carbonic anhydrase to accelerate carbon dioxide capture processes. J. Chem. Technol. Biotechnol. 2015, 90, 3–10. [Google Scholar] [CrossRef]
- Power, I.M.; Harrison, A.L.; Dipple, G.M. Accelerating Mineral Carbonation Using Carbonic Anhydrase. Environ. Sci. Technol. 2016, 50, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Merle, G.; Fradette, S.; Madore, E.; Barralet, J.E. Electropolymerized Carbonic Anhydrase Immobilization for Carbon Dioxide Capture. Langmuir 2014, 30, 6915–6919. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.J.; Weber, H.; Bhattacharyya, P.; Argyle, M.; Taylor, D.; Christensen, M.; Foulke, T.; Fahlsing, P. Instantaneous Capture and Mineralization of Flue Gas Carbon Dioxide: Pilot Scale Study. Nat. Proc. 2010. [Google Scholar] [CrossRef]
- Alvizo, O.; Nguyen, L.J.; Savile, C.K.; Bresson, J.A.; Lakhapatri, S.L.; Solis, E.O.P.; Fox, R.J.; Broering, J.M.; Benoit, M.R.; Zimmerman, S.A.; et al. Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas. Proc. Natl. Acad. Sci. USA 2014, 111, 16436–16441. [Google Scholar] [CrossRef] [Green Version]
- Bose, H.; Satyanarayana, T. Microbial Carbonic Anhydrases in Biomimetic Carbon Sequestration for Mitigating Global Warming: Prospects and Perspectives. Front. Microbiol. 2017, 8, 1615. [Google Scholar] [CrossRef]
- Kelsey, R.A.; Miller, D.A.; Parkin, S.R.; Liu, K.; Remias, J.E.; Yang, Y.; Lightstone, F.C.; Liu, K.; Lippert, C.A.; Odom, S.A. Carbonic anhydrase mimics for enhanced CO2 absorption in an amine-based capture solvent. Dalton Trans. 2016, 45, 324–333. [Google Scholar] [CrossRef]
- Larachi, F. Kinetic Model for the Reversible Hydration of Carbon Dioxide Catalyzed by Human Carbonic Anhydrase II. Ind. Eng. Chem. Res. 2010, 49, 9095–9104. [Google Scholar] [CrossRef]
- Eriksson, A.E.; Jones, T.A.; Liljas, A. Refined structure of human carbonic anhydrase II at 2.0 Å resolution. Proteins Struct. Funct. Bioinform. 1988, 4, 274–282. [Google Scholar] [CrossRef]
- Xue, Y.; Liljas, A.; Jonsson, B.-H.; Lindskog, S. Structural analysis of the zinc hydroxide–Thr-199–Glu-106 hydrogen-bond network in human carbonic anhydrase II. Proteins Struct. Funct. Bioinform. 1993, 17, 93–106. [Google Scholar] [CrossRef]
- Ferraroni, M.; Tilli, S.; Briganti, F.; Chegwidden, W.R.; Supuran, C.T.; Wiebauer, K.E.; Tashian, R.E.; Scozzafava, A. Crystal Structure of a Zinc-Activated Variant of Human Carbonic Anhydrase I, CA I Michigan 1: Evidence for a Second Zinc Binding Site Involving Arginine Coordination. Biochemistry 2002, 41, 6237–6244. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, J.A.; Christianson, D.W. Structure of an engineered His3 Cys zinc binding site in human carbonic anhydrase II. Biochemistry 1993, 32, 9901–9905. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, J.A.; Baird, T.T.; McGee, S.A.; Christianson, D.W.; Fierke, C.A. Structure-assisted redesign of a protein-zinc-binding site with femtomolar affinity. Proc. Natl. Acad. Sci. USA 1995, 92, 5017–5021. [Google Scholar] [CrossRef] [PubMed]
- Floyd, W.C.; Baker, S.E.; Valdez, C.A.; Stolaroff, J.K.; Bearinger, J.P.; Satcher, J.H.; Aines, R.D. Evaluation of a Carbonic Anhydrase Mimic for Industrial Carbon Capture. Environ. Sci. Technol. 2013, 47, 10049–10055. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [CrossRef]
- Galardon, E.; Tomas, A.; Roussel, P.; Artaud, I. Synthesis, Stability, and Reactivity of [(TPA)Zn(SH)]+ in Aqueous and Organic Solutions. Eur. J. Inorg. Chem. 2011, 2011, 3797–3801. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, J.; Zhu, L.; Duarandin, A.; G, Y.V.; Geacintov, N.; Canary, J.W. Structures, Metal Ion Affinities, and Fluorescence Properties of Soluble Derivatives of Tris((6-phenyl-2-pyridyl)methyl)amine. Inorg. Chem. 2009, 48, 11196–11208. [Google Scholar] [CrossRef] [Green Version]
- Mareque-Rivas, J.C.; Prabaharan, R.; Parsons, S. Quantifying the relative contribution of hydrogen bonding and hydrophobic environments, and coordinating groups, in the zinc(II)–water acidity by synthetic modelling chemistry. Dalton Trans. 2004, 1648–1655. [Google Scholar] [CrossRef]
- Spiropulos, N.G.; Standley, E.A.; Shaw, I.R.; Ingalls, B.L.; Diebels, B.; Krawczyk, S.V.; Gherman, B.F.; Arif, A.M.; Brown, E.C. Synthesis of zinc and cadmium O-alkyl thiocarbonate and dithiocarbonate complexes and a cationic zinc hydrosulfide complex. Inorg. Chim. Acta 2012, 386, 83–92. [Google Scholar] [CrossRef]
- Koziol, L.; Valdez, C.A.; Baker, S.E.; Lau, E.Y.; Floyd, W.C.; Wong, S.E.; Satcher, J.H.; Lightstone, F.C.; Aines, R.D. Toward a Small Molecule, Biomimetic Carbonic Anhydrase Model: Theoretical and Experimental Investigations of a Panel of Zinc(II) Aza-Macrocyclic Catalysts. Inorg. Chem. 2012, 51, 6803–6812. [Google Scholar] [CrossRef]
- Nakata, K.; Shimomura, N.; Shiina, N.; Izumi, M.; Ichikawa, K.; Shiro, M. Kinetic study of catalytic CO2 hydration by water-soluble model compound of carbonic anhydrase and anion inhibition effect on CO2 hydration. J. Inorg. Biochem. 2002, 89, 255–266. [Google Scholar] [CrossRef]
- Park, D.; Lee, M.S. Kinetic study of catalytic CO2 hydration by metal-substituted biomimetic carbonic anhydrase model complexes. R. Soc. Open Sci. 2019, 6, 190407. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Maupin, C.M.; McKenna, R.; Silverman, D.N.; Voth, G.A. Elucidation of the Proton Transport Mechanism in Human Carbonic Anhydrase II. J. Am. Chem. Soc. 2009, 131, 7598–7608. [Google Scholar] [CrossRef] [Green Version]
- Silverman, D.N.; Lindskog, S. The catalytic mechanism of carbonic anhydrase: Implications of a rate-limiting protolysis of water. Acc. Chem. Res. 1988, 21, 30–36. [Google Scholar] [CrossRef]
- Rains, J.G.D.; O’Donnelly, K.; Oliver, T.; Woscholski, R.; Long, N.J.; Barter, L.M.C. Bicarbonate Inhibition of Carbonic Anhydrase Mimics Hinders Catalytic Efficiency: Elucidating the Mechanism and Gaining Insight toward Improving Speed and Efficiency. ACS Catal. 2019, 9, 1353–1365. [Google Scholar] [CrossRef]
- Roque-Malherbe, R.; Lugo, F.; Polanco, R. Synthesis, structural elucidation and carbon dioxide adsorption on Zn (II) hexacyanoferrate (II) Prussian blue analogue. Appl. Surf. Sci. 2016, 385, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Kanari, N.; Mishra, D.; Gaballah, I.; Dupré, B. Thermal decomposition of zinc carbonate hydroxide. Thermochim. Acta 2004, 410, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Berreau, L.M.; Makowska-Grzyska, M.M.; Arif, A.M. Modeling the Active Site Chemistry of Liver Alcohol Dehydrogenase: Mononuclear Zinc Methanol and N,N-Dimethylformamide Complexes of a Nitrogen/Sulfur Ligand Possessing an Internal Hydrogen Bond Donor. Inorg. Chem. 2001, 40, 2212–2213. [Google Scholar] [CrossRef]
- Koziol, L.; Essiz, S.G.; Wong, S.E.; Lau, E.Y.; Valdez, C.A.; Satcher, J.H.; Aines, R.D.; Lightstone, F.C. Computational Analysis of a Zn-Bound Tris(imidazolyl) Calix[6]arene Aqua Complex: Toward Incorporating Second-Coordination Sphere Effects into Carbonic Anhydrase Biomimetics. J. Chem. Theory Comput. 2013, 9, 1320–1327. [Google Scholar] [CrossRef]
- Kiefer, L.L.; Paterno, S.A.; Fierke, C.A. Hydrogen bond network in the metal binding site of carbonic anhydrase enhances zinc affinity and catalytic efficiency. J. Am. Chem. Soc. 1995, 117, 6831–6837. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, L.C.; Mboge, Y.M.; Frost, C.S.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [PubMed]
- Genis, C.; Sippel, K.H.; Case, N.; Cao, W.; Avvaru, B.S.; Tartaglia, L.J.; Govindasamy, L.; Tu, C.; Agbandje-McKenna, M.; Silverman, D.N.; et al. Design of a Carbonic Anhydrase IX Active-Site Mimic to Screen Inhibitors for Possible Anticancer Properties. Biochemistry 2009, 48, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.F.; Ippolito, J.A.; Christianson, D.W.; Fierke, C.A. Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II. J. Biol. Chem. 1993, 268, 27458–27466. [Google Scholar]
- Ma, R.; Schuette, G.F.; Broadbelt, L.J. Insights into the Relationship of Catalytic Activity and Structure: A Comparison Study of Three Carbonic Anhydrase Mimics. Int. J. Chem. Kinet. 2014, 46, 683–700. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | (1) | (2) |
|---|---|---|
| pKa | 8.0 | 6.8 |
| Complexes | kobs | kind | σobs |
|---|---|---|---|
| (1) | 648.4 | 717.88 | 23.7 |
| (2) | 730.6 | 735.21 | 53.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.; Lee, M.S. Kinetic Study of CO2 Hydration by Small-Molecule Catalysts with A Second Coordination Sphere that Mimic the Effect of the Thr-199 Residue of Carbonic Anhydrase. Biomimetics 2019, 4, 66. https://doi.org/10.3390/biomimetics4040066
Park D, Lee MS. Kinetic Study of CO2 Hydration by Small-Molecule Catalysts with A Second Coordination Sphere that Mimic the Effect of the Thr-199 Residue of Carbonic Anhydrase. Biomimetics. 2019; 4(4):66. https://doi.org/10.3390/biomimetics4040066
Chicago/Turabian StylePark, DongKook, and Man Sig Lee. 2019. "Kinetic Study of CO2 Hydration by Small-Molecule Catalysts with A Second Coordination Sphere that Mimic the Effect of the Thr-199 Residue of Carbonic Anhydrase" Biomimetics 4, no. 4: 66. https://doi.org/10.3390/biomimetics4040066
APA StylePark, D., & Lee, M. S. (2019). Kinetic Study of CO2 Hydration by Small-Molecule Catalysts with A Second Coordination Sphere that Mimic the Effect of the Thr-199 Residue of Carbonic Anhydrase. Biomimetics, 4(4), 66. https://doi.org/10.3390/biomimetics4040066