


A Novel BC2N Monolayer as Anode Material for Li-Ion Battery


Abstract
1. Introduction
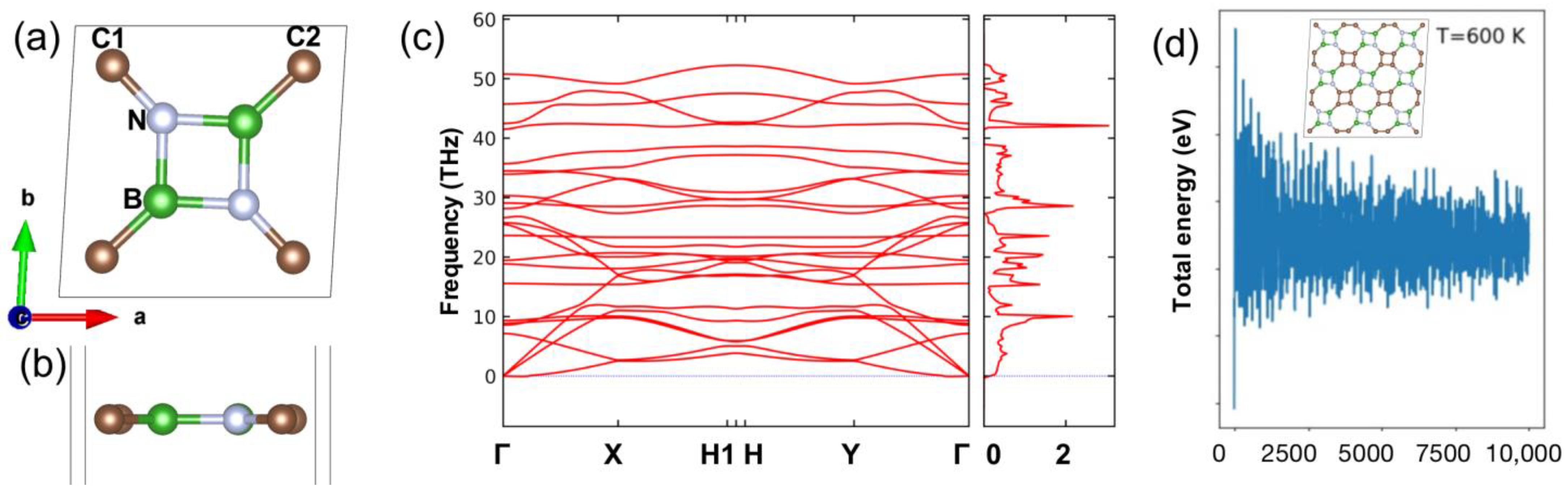
2. Stability of BC2N Monolayer
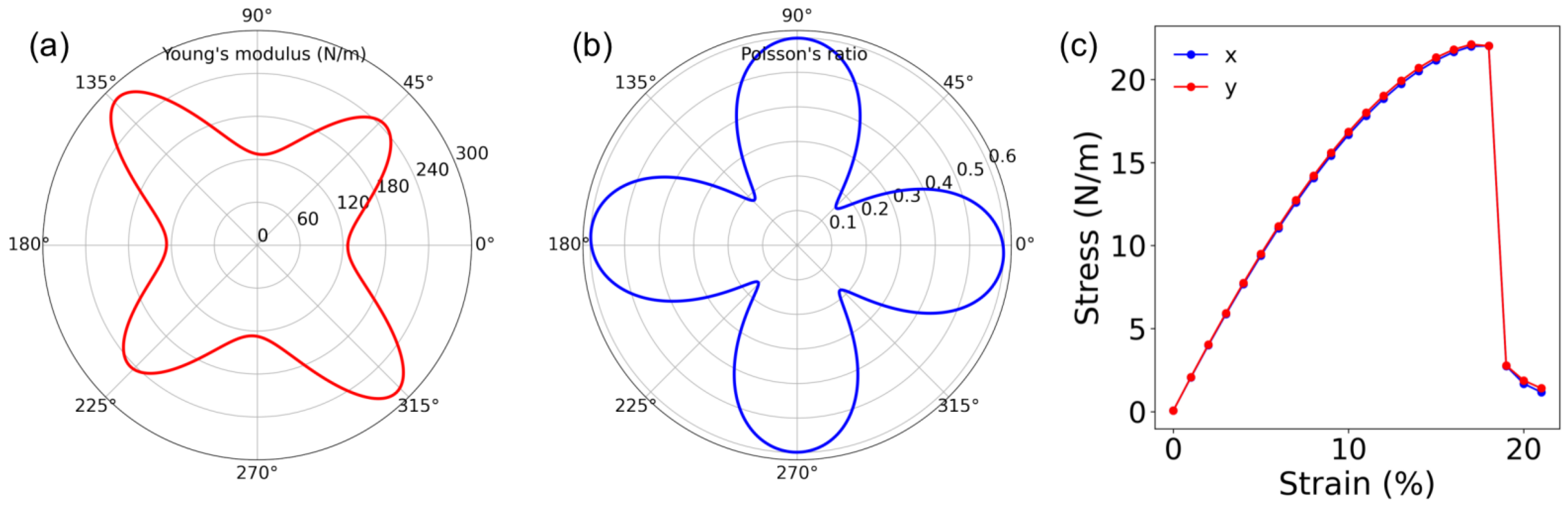
3. Mechanical Properties
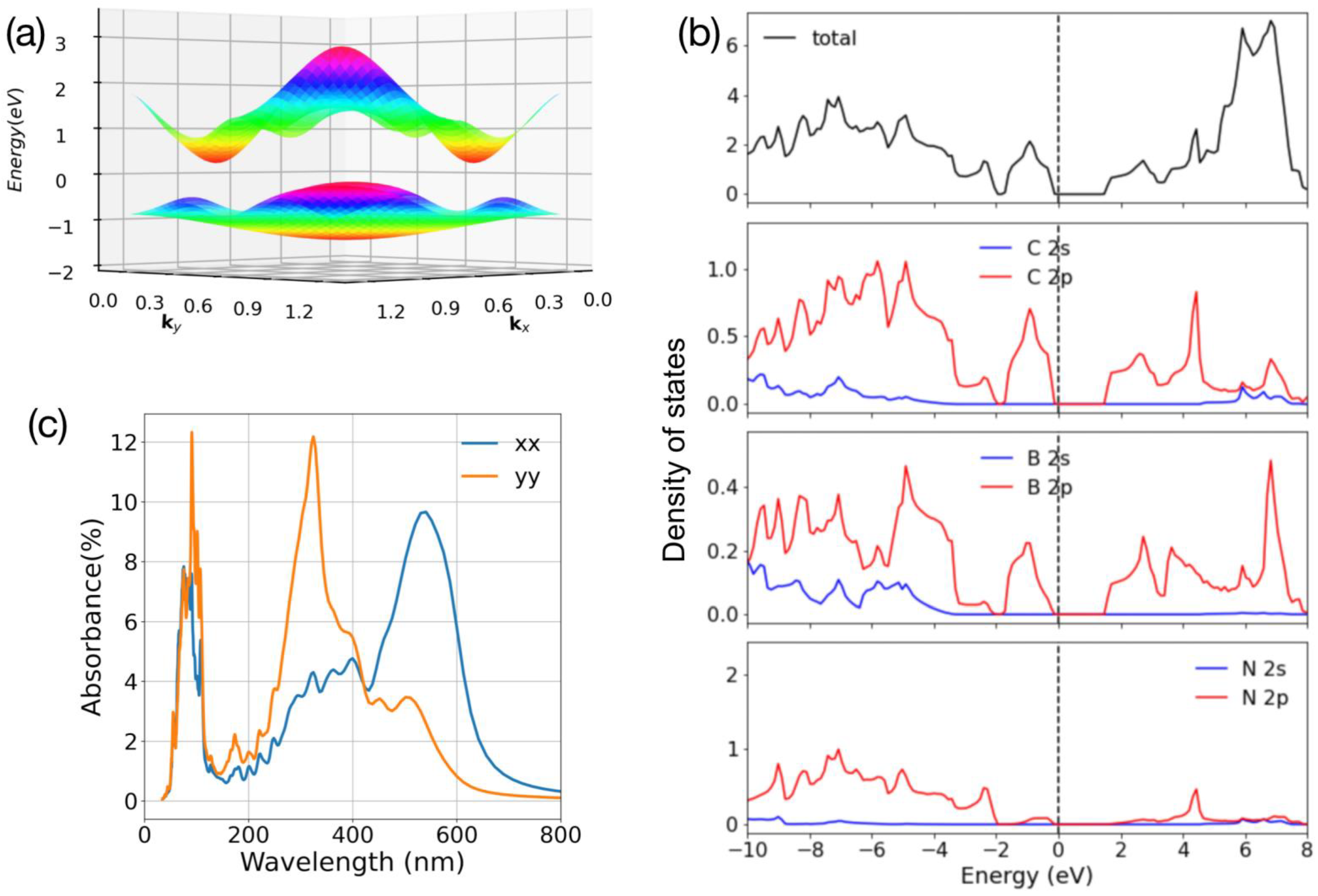
4. Electronic and Optical Properties
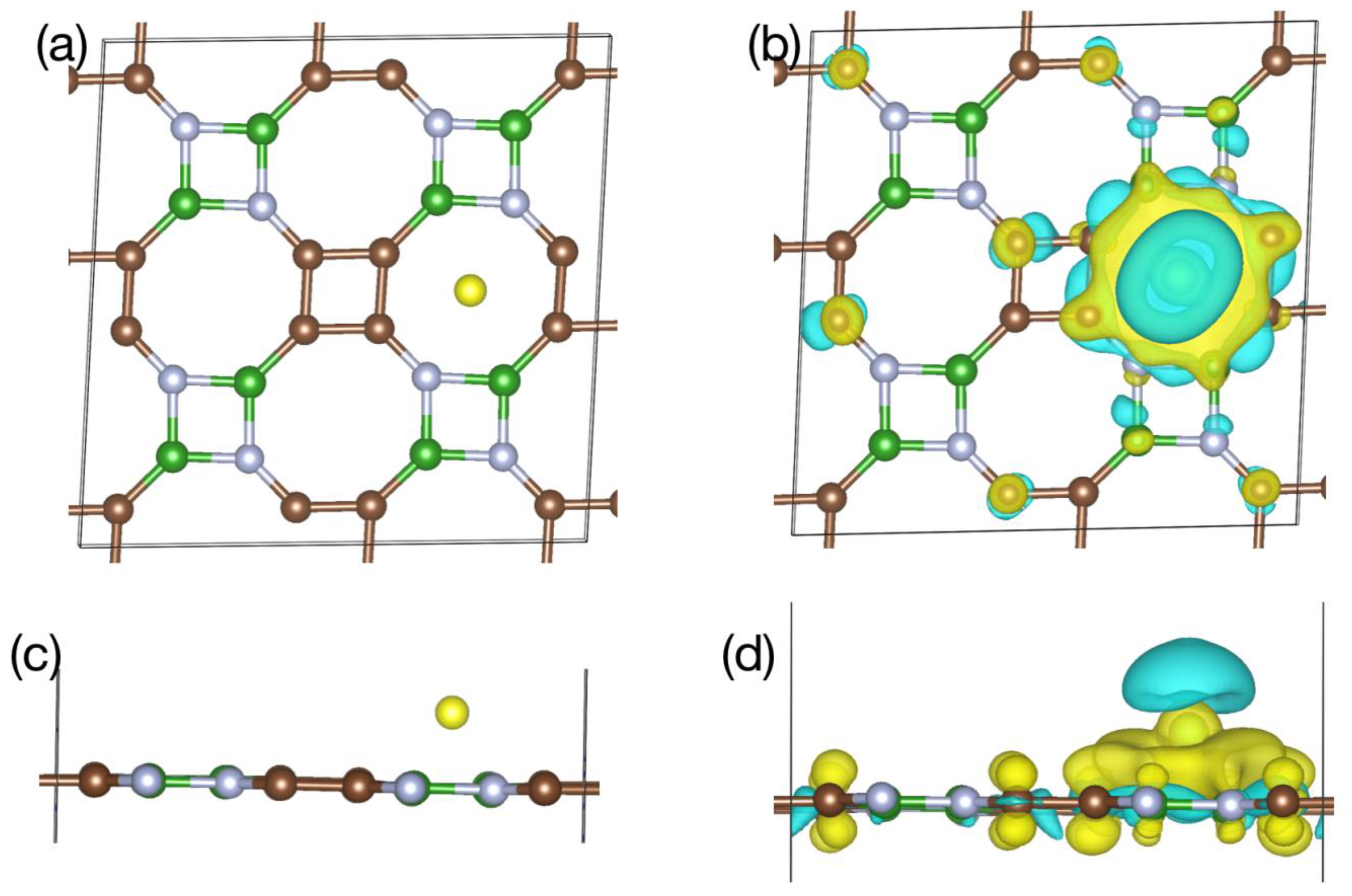
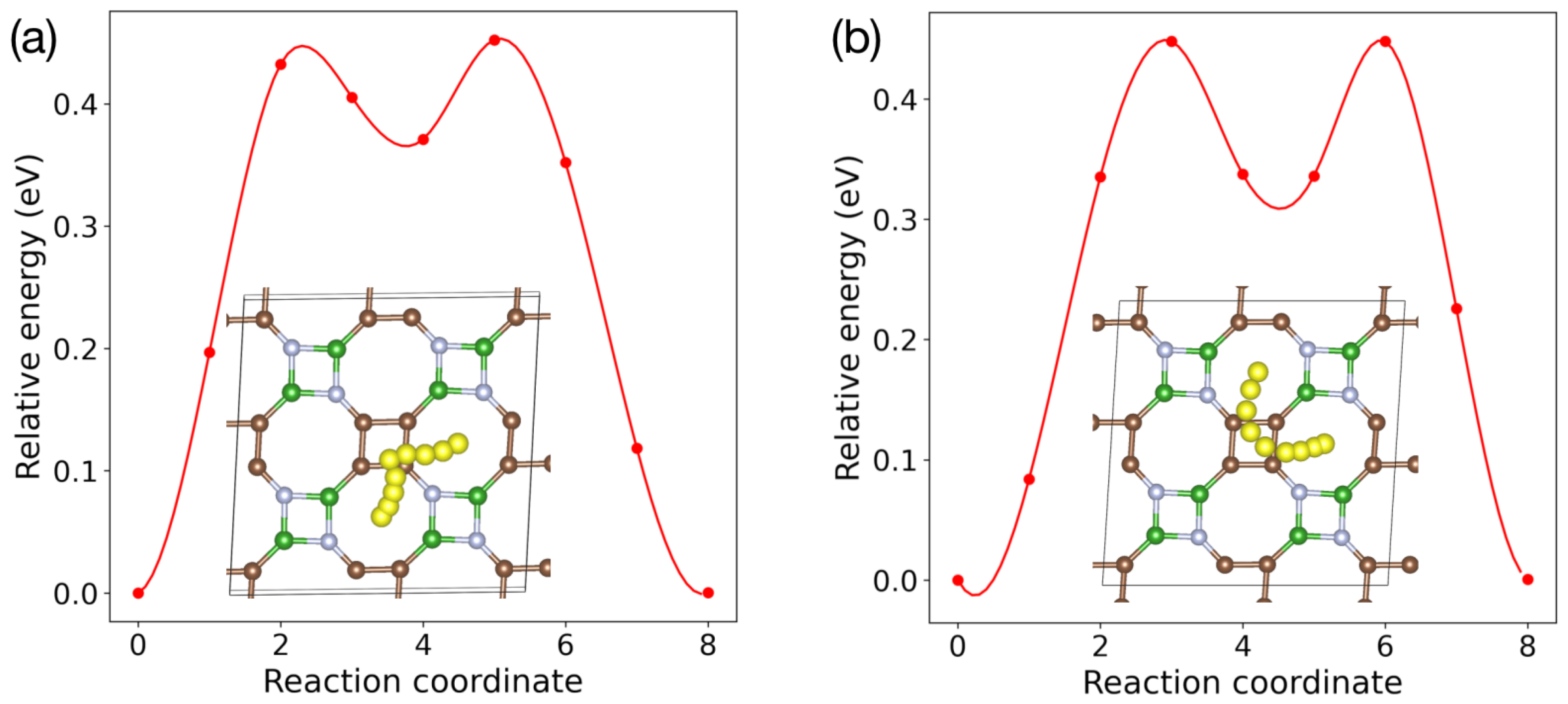
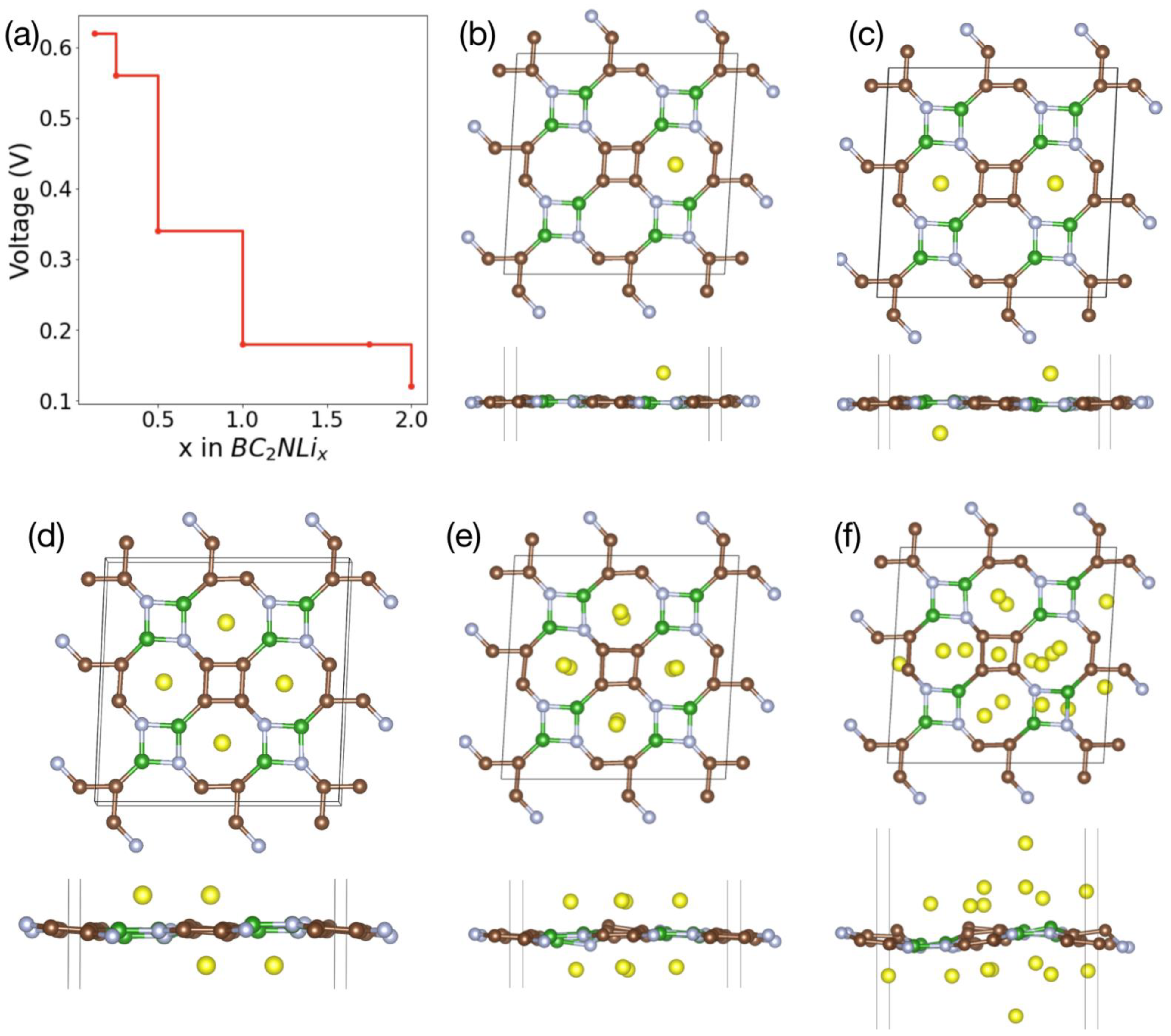
5. Li Storage Properties
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olabi, A.G.; Abdelkareem, M.A.; Wilberforce, T.; Sayed, E.T. Application of graphene in energy storage device—A review. Renew. Sustain. Energy Rev. 2021, 135, 110026. [Google Scholar] [CrossRef]
- Huang, H.; Shi, H.; Das, P.; Qin, J.; Li, Y.; Wang, X.; Su, F.; Wen, P.; Li, S.; Lu, P. The chemistry and promising applications of graphene and porous graphene materials. Adv. Funct. Mater. 2020, 30, 1909035. [Google Scholar] [CrossRef]
- Rojaee, R.; Shahbazian-Yassar, R. Two-dimensional materials to address the lithium battery challenges. ACS Nano 2020, 14, 2628–2658. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Sun, G.; Hu, L.; Xiao, Y.; Zhang, Z.; Qu, L. Recent progress in graphene-based electrodes for flexible batteries. InfoMat 2020, 2, 509–526. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, Y.; Yan, X.; Cohen, M.L.; Zhang, S. Topological carbon materials: A new perspective. Phys. Rep. 2020, 868, 1–32. [Google Scholar] [CrossRef]
- Nehate, S.D.; Saikumar, A.K.; Prakash, A.; Sundaram, K.B. A review of boron carbon nitride thin films and progress in nanomaterials. Mater. Today Adv. 2020, 8, 100106. [Google Scholar] [CrossRef]
- Kida, T.; Shigezumi, K.; Mannan, M.A.; Akiyama, M.; Baba, Y.; Nagano, M. Synthesis of boron carbonitride (BCN) films by plasma-enhanced chemical vapor deposition using trimethylamine borane as a molecular precursor. Vacuum 2009, 83, 1143–1146. [Google Scholar] [CrossRef]
- Ci, L.; Song, L.; Jin, C.; Jariwala, D.; Wu, D.; Li, Y.; Srivastava, A.; Wang, Z.F.; Storr, K.; Balicas, L.; et al. Atomic layers of hybridized boron nitride and graphene domains. Nat. Mater. 2010, 9, 430–435. [Google Scholar] [CrossRef]
- Huang, C.; Chen, C.; Zhang, M.; Lin, L.; Ye, X.; Lin, S.; Antonietti, M.; Wang, X. Carbon-doped BN nanosheets for metal-free photoredox catalysis. Nat. Commun. 2015, 6, 7698. [Google Scholar] [CrossRef]
- Zhang, K.; Feng, Y.; Wang, F.; Yang, Z.; Wang, J. Two dimensional hexagonal boron nitride (2D-hBN): Synthesis, properties and applications. J. Mater. Chem. C 2017, 5, 11992–12022. [Google Scholar] [CrossRef]
- Chhetri, M.; Maitra, S.; Chakraborty, H.; Waghmare, U.V.; Rao, C. Superior performance of borocarbonitrides, B x C y N z, as stable, low-cost metal-free electrocatalysts for the hydrogen evolution reaction. Energy Environ. Sci. 2016, 9, 95–101. [Google Scholar] [CrossRef]
- Fiori, G.; Betti, A.; Bruzzone, S.; Iannaccone, G. Lateral graphene–hBCN heterostructures as a platform for fully two-dimensional transistors. ACS Nano 2012, 6, 2642–2648. [Google Scholar] [CrossRef]
- Beniwal, S.; Hooper, J.; Miller, D.P.; Costa, P.S.; Chen, G.; Liu, S.Y.; Dowben, P.A.; Sykes, E.C.; Zurek, E.; Enders, A. Graphene-like Boron-Carbon-Nitrogen Monolayers. ACS Nano 2017, 11, 2486–2493. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Gopalakrishnan, K. Borocarbonitrides, B(x)C(y)N(z): Synthesis, Characterization, and Properties with Potential Applications. ACS Appl. Mater. Interfaces 2017, 9, 19478–19494. [Google Scholar] [CrossRef] [PubMed]
- Mou, P.; Zhao, J.; Wang, G.; Shi, S.; Wan, G.; Zhou, M.; Deng, Z.; Teng, S.; Wang, G. BCN nanosheets derived from coconut shells with outstanding microwave absorption and thermal conductive properties. Chem. Eng. J. 2022, 437, 135285. [Google Scholar] [CrossRef]
- Hasan, M.M.; Khedr, G.E.; Zakaria, F.; Allam, N.K. Intermolecular electron transfer in electrochemically exfoliated BCN-Cu nanosheet electrocatalysts for efficient hydrogen evolution. ACS Appl. Energy Mater. 2022, 5, 9692–9701. [Google Scholar] [CrossRef]
- Kaur, M.; Singh, K.; Vij, A.; Kumar, A. Recent insights into BCN nanomaterials—Synthesis, properties and applications. New J. Chem. 2023, 47, 2137–2160. [Google Scholar] [CrossRef]
- Seo, T.H.; Lee, W.; Lee, K.S.; Hwang, J.Y.; Son, D.I.; Ahn, S.; Cho, H.; Kim, M.J. Dominant formation of h-BC2N in h-BxCyNz films: CVD synthesis and characterization. Carbon 2021, 182, 791–798. [Google Scholar] [CrossRef]
- Lam, K.-T.; Lu, Y.; Feng, Y.P.; Liang, G. Stability and electronic structure of two dimensional Cx(BN)y compound. Appl. Phys. Lett. 2011, 98, 022101. [Google Scholar] [CrossRef]
- Bafekry, A.; Naseri, M.; Fadlallah, M.M.; Abdolhosseini Sarsari, I.; Faraji, M.; Bagheri Khatibani, A.; Ghergherehchi, M.; Gogova, D. A novel two-dimensional boron–carbon–nitride (BCN) monolayer: A first-principles insight. J. Appl. Phys. 2021, 130, 114301. [Google Scholar] [CrossRef]
- Zhang, M.; Gao, G.; Kutana, A.; Wang, Y.; Zou, X.; Tse, J.S.; Yakobson, B.I.; Li, H.; Liu, H.; Ma, Y. Two-dimensional boron-nitrogen-carbon monolayers with tunable direct band gaps. Nanoscale 2015, 7, 12023–12029. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, X.-F.; Chen, J.; Fan, Y.-X.; Wang, H.-T.; Guo, X.; He, J.; Tian, Y. Infrared and Raman spectra of β−BC2N from first principles calculations. Phys. Rev. B 2006, 74, 193101. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, X.; Wang, M. Theoretical investigations of a new two-dimensional semiconducting boron-carbon-nitrogen structure. RSC Adv. 2020, 10, 3424–3428. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, R.C.; Guimarães, P.S.; Baierle, R.J. First principles study of native defects in a graphitic BC2N monolayer. Thin Solid Film. 2010, 518, 4356–4362. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, Z.Y.; Yang, D.Z.; Xue, D.S.; Si, M.S. Theoretical Prediction of Carrier Mobility in Few-Layer BC2N. J. Phys. Chem. Lett. 2014, 5, 4073–4077. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Meng, X.; Zhou, S.; Yang, G.; Zhou, X. Isoelectronic analogues of graphene: The BCN monolayers with visible-light absorption and high carrier mobility. J. Phys. Condens. Matter 2019, 31, 125301. [Google Scholar] [CrossRef]
- Alyörük, M.M. Piezoelectricity in monolayer BxCyNz structures: A first principles study. Comput. Mater. Sci. 2021, 195, 110505. [Google Scholar] [CrossRef]
- Bafekry, A.; Naseri, M.; Faraji, M.; Fadlallah, M.M.; Hoat, D.M.; Jappor, H.R.; Ghergherehchi, M.; Gogova, D.; Afarideh, H. Theoretical prediction of two-dimensional BC2X (X = N, P, As) monolayers: Ab initio investigations. Sci. Rep. 2022, 12, 22269. [Google Scholar] [CrossRef]
- Yu, J.; He, C.; Huo, J.; Zhao, C.; Yu, L. Electric field controlled CO2 capture and activation on BC6N monolayers: A first-principles study. Surf. Interfaces 2022, 30, 101885. [Google Scholar] [CrossRef]
- Chen, L.; Yang, M.; Kong, F.; Du, W.; Guo, J.; Shu, H. Penta-BCN monolayer with high specific capacity and mobility as a compelling anode material for rechargeable batteries. Phys. Chem. Chem. Phys. 2021, 23, 17693–17702. [Google Scholar] [CrossRef]
- Kilic, M.E.; Lee, K.-R. Emerging exotic properties of two-dimensional ternary tetrahexagonal BCN: Tunable anisotropic transport properties with huge excitonic effects for nanoelectronics and optoelectronics. Mater. Today Phys. 2022, 27, 100792. [Google Scholar] [CrossRef]
- Kilic, M.E.; Lee, K.-R. Two-Dimensional Ternary Pentagonal BCN: A Promising Photocatalyst Semiconductor for Water Splitting with Strong Excitonic Effects. Phys. Rev. Appl. 2022, 18, 014066. [Google Scholar] [CrossRef]
- Dabsamut, K.; Thanasarnsurapong, T.; Maluangnont, T.; Jiraroj, T.; Jungthawan, S.; Boonchun, A. Strain engineering and thermal conductivity of a penta-BCN monolayer: A computational study. J. Phys. D Appl. Phys. 2021, 54, 355301. [Google Scholar] [CrossRef]
- Matar, S.F.; Solozhenko, V.L. New superhard tetragonal BCN from crystal chemistry and first principles. Materialia 2022, 26, 101581. [Google Scholar] [CrossRef]
- Ananchuensook, A.; Dabsamut, K.; Thanasarnsurapong, T.; Maluangnont, T.; Jiraroj, T.; Jungthawan, S.; Boonchun, A. Towards a new packing pattern of Li adsorption in two-dimensional pentagonal BCN. Phys. Chem. Chem. Phys. 2022, 24, 13194–13200. [Google Scholar] [CrossRef]
- Majidi, R. Electronic properties of T graphene-like C–BN sheets: A density functional theory study. Phys. E Low-Dimens. Syst. Nanostruct. 2015, 74, 371–376. [Google Scholar] [CrossRef]
- Adekoya, D.; Qian, S.; Gu, X.; Wen, W.; Li, D.; Ma, J.; Zhang, S. DFT-guided design and fabrication of carbon-nitride-based materials for energy storage devices: A review. Nano Micro Lett. 2021, 13, 13. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Ji, H.; Zhou, J.; Wang, Z.; Qian, T.; Yan, C. Boosting the optimization of lithium metal batteries by molecular dynamics simulations: A perspective. Adv. Energy Mater. 2020, 10, 2002373. [Google Scholar] [CrossRef]
- He, B.; Wang, Y.; Zhai, Q.; Qiu, P.; Dong, G.; Liu, X.; Chen, Y.; Li, Z. From polymeric carbon nitride to carbon materials: Extended application to electrochemical energy conversion and storage. Nanoscale 2020, 12, 8636–8646. [Google Scholar] [CrossRef]
- Chen, J.; Mao, Z.; Zhang, L.; Wang, D.; Xu, R.; Bie, L.; Fahlman, B.D. Nitrogen-deficient graphitic carbon nitride with enhanced performance for lithium ion battery anodes. ACS Nano 2017, 11, 12650–12657. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic coefficients from ab initio total energy calculations. Phys. Rev. B 2001, 63, 174103. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Kudin, K.N.; Scuseria, G.E.; Yakobson, B.I. C 2 F, BN, and C nanoshell elasticity from ab initio computations. Phys. Rev. B 2001, 64, 235406. [Google Scholar] [CrossRef]
- Wang, B.; Wu, Q.; Zhang, Y.; Ma, L.; Wang, J. Auxetic B(4)N Monolayer: A Promising 2D Material with in-Plane Negative Poisson’s Ratio and Large Anisotropic Mechanics. ACS Appl. Mater. Interfaces 2019, 11, 33231–33237. [Google Scholar] [CrossRef]
- Wang, S.; Yang, B.; Chen, H.; Ruckenstein, E. Reconfiguring graphene for high-performance metal-ion battery anodes. Energy Storage Mater. 2019, 16, 619–624. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, J.; Cheng, Y.; Yang, H.Y.; Yao, Y.; Yang, S.A. Borophene as an extremely high capacity electrode material for Li-ion and Na-ion batteries. Nanoscale 2016, 8, 15340–15347. [Google Scholar] [CrossRef]
- Wu, D.; Yang, B.; Chen, H.; Ruckenstein, E. Nitrogenated holey graphene C2N monolayer anodes for lithium-and sodium-ion batteries with high performance. Energy Storage Mater. 2019, 16, 574–580. [Google Scholar] [CrossRef]
- Wang, S.; Si, Y.; Yang, B.; Ruckenstein, E.; Chen, H. Two-dimensional carbon-based auxetic materials for broad-spectrum metal-ion battery anodes. J. Phys. Chem. Lett. 2019, 10, 3269–3275. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized Gradient Approximation for the Exchange-Correlation Hole of a Many-Electron System. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (Dft-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, I.C.; Ángyán, J.G. Screened Hybrid Density Functionals Applied to Solids. J. Chem. Phys. 2006, 124, 154709. [Google Scholar] [CrossRef] [PubMed]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic Zro2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C11 (J/m2) | C12 (J/m2) | C22 (J/m2) | C26 (J/m2) | C66 (J/m2) | Yx (N/m) | Yy (N/m) | νx | νy | |
|---|---|---|---|---|---|---|---|---|---|
| BC2N | 198 | 119 | 200 | −14 | 113 | 127 | 127 | 0.60 | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Lin, J.; Lin, Q.; Li, R.; He, H. A Novel BC2N Monolayer as Anode Material for Li-Ion Battery. Batteries 2023, 9, 315. https://doi.org/10.3390/batteries9060315
Chen X, Lin J, Lin Q, Li R, He H. A Novel BC2N Monolayer as Anode Material for Li-Ion Battery. Batteries. 2023; 9(6):315. https://doi.org/10.3390/batteries9060315
Chicago/Turabian StyleChen, Xiaowei, Jiahe Lin, Qiubao Lin, Renquan Li, and Hongsheng He. 2023. "A Novel BC2N Monolayer as Anode Material for Li-Ion Battery" Batteries 9, no. 6: 315. https://doi.org/10.3390/batteries9060315
APA StyleChen, X., Lin, J., Lin, Q., Li, R., & He, H. (2023). A Novel BC2N Monolayer as Anode Material for Li-Ion Battery. Batteries, 9(6), 315. https://doi.org/10.3390/batteries9060315