


Solid–Electrolyte Interface Formation on Si Nanowires in Li-Ion Batteries: The Impact of Electrolyte Additives


 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Silicon Nanowires (Si NWs): Direct Electrode Manufacture
2.2. Cell formulation and Electrochemical Test Conditions
2.3. Scanning Electron Microscopy
2.4. Raman Micro-Spectroscopy
2.5. FTIR-ATR Spectroscopy
2.6. Optical-Photothermal Infrared Spectroscopy
3. Results
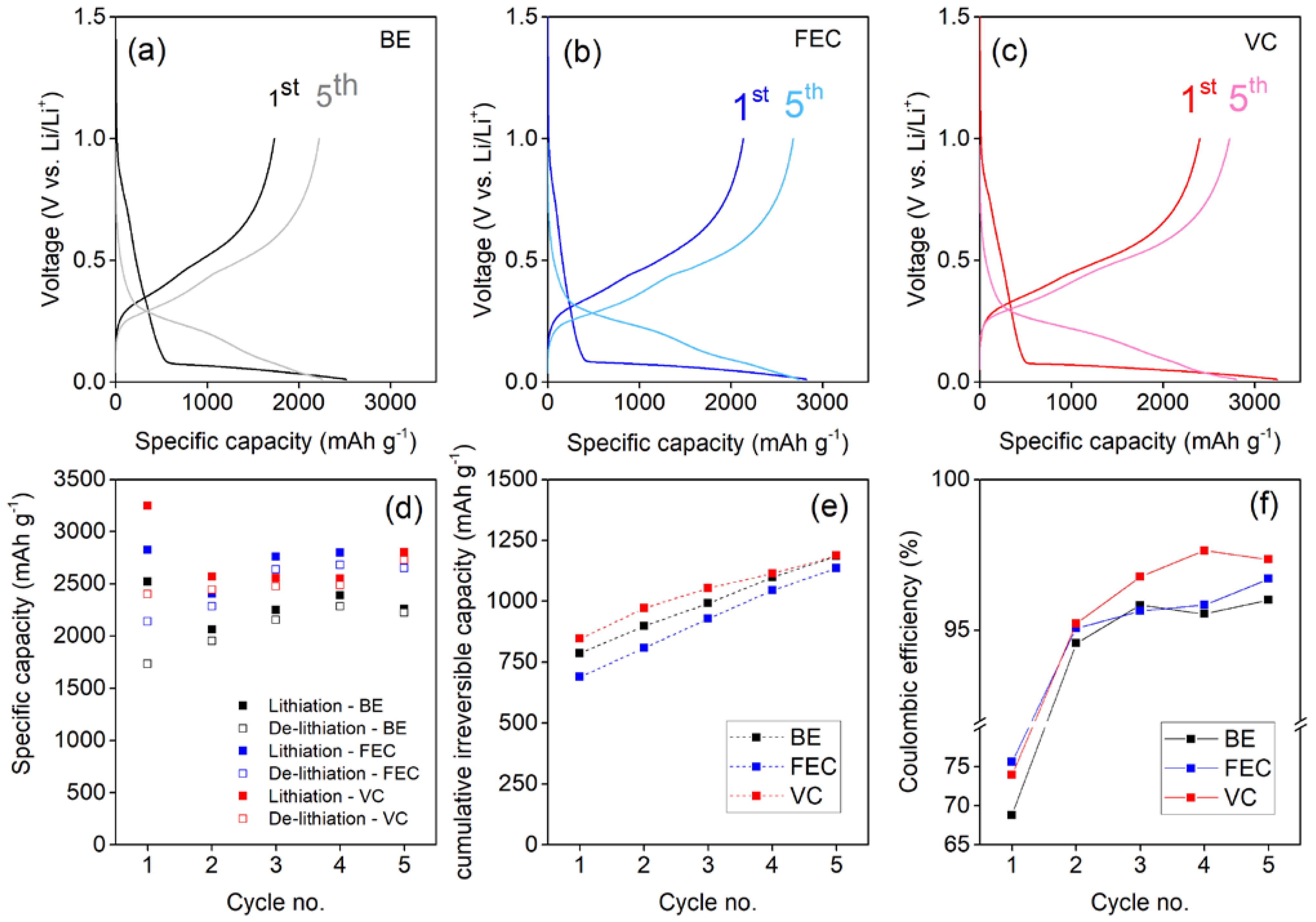
3.1. Electrochemical Formation and Consolidation of the SEI
3.2. Morphological Characterization
3.3. Micro-Spectroscopic Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Martinho, F. Challenges for the Future of Tandem Photovoltaics on the Path to Terawatt Levels: A Technology Review. Energy Environ. Sci. 2021, 14, 3840–3871. [Google Scholar] [CrossRef]
- Marri, I.; Ossicini, S. Multiple Exciton Generation in Isolated and Interacting Silicon Nanocrystals. Nanoscale 2021, 13, 12119–12142. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Dong, D.; Wang, J.; Liu, D.; Yu, X.; Zheng, Y.; Wen, Z.; Lei, W.; Deng, Y.; Wang, J.; et al. Silicon-Based Lithium Ion Battery Systems: State-of-the-Art from Half and Full Cell Viewpoint. Adv. Funct. Mater. 2021, 31, 2102546. [Google Scholar] [CrossRef]
- Sarkar, A.; Lee, Y.; Ahn, J.-H. Si Nanomebranes: Material Properties and Applications. Nano Res. 2021, 14, 3010–3032. [Google Scholar] [CrossRef]
- Liu, C.; Guo, J.; Yu, L.; Li, J.; Zhang, M.; Li, H.; Shi, Y.; Dai, D. Silicon/2D-Material Photodetectors: From near-Infrared to Mid-Infrared. Light Sci. Appl. 2021, 10, 123. [Google Scholar] [CrossRef]
- Siffert, P.; Krimmel, E.F. Silicon: Evolution and Future of a Technology; Springer: Berlin, Germany, 2004; p. 549. [Google Scholar]
- Kim, H.; Seo, M.; Park, M.H.; Cho, J. A Critical Size of Silicon Nano-Anodes for Lithium Rechargeable Batteries. Angew. Chim. Int. Ed. 2010, 49, 2146–2149. [Google Scholar] [CrossRef]
- Su, X.; Wu, Q.; Li, J.; Xiao, X.; Lott, A.; Lu, W.; Sheldon, B.W.; Wu, J. Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review. Adv. Energy Mater. 2014, 4, 1300882. [Google Scholar] [CrossRef]
- Liu, J.; Kopold, P.; van Aken, P.A.; Maier, J.; Yu, Y. Energy Storage Materials from Nature through Nanotechnology: ASustainable Route from Reed Plants to a Silicon Anode for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2015, 127, 9768–9772. [Google Scholar] [CrossRef]
- Jin, Y.; Zhu, B.; Lu, Z.; Liu, N.; Zhu, J. Challenges and Recent Progress in the Development of Si Anodes for Lithium-Ion Battery. Adv. Energy Mater. 2017, 7, 1700715. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C.; Dou, Y.; Cheng, N.; Cui, D.; Du, Y.; Liu, P.; Al-Mamun, M.; Zhang, S.; Zhao, H. A Yolk–Shell Structured Silicon Anode with Superior Conductivity and High Tap Density for Full Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2019, 58, 8824–8828. [Google Scholar] [CrossRef]
- Su, H.; Barragan, A.A.; Geng, L.; Long, D.; Ling, L.; Bozhilov, K.N.; Mangolini, L.; Guo, J. Colloidal Synthesis of Silicon–Carbon Composite Material for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2017, 56, 10780–10785. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Ren, Y.; Mu, T.; Yin, X.; Du, C.; Huo, H.; Cheng, X.; Zuo, P.; Yin, G. Engineering Molecular Polymerization for Template-Free SiOx/C Hollow Spheres as Ultrastable Anodes in Lithium-Ion Batteries. Adv. Funct. Mater. 2021, 31, 2101145. [Google Scholar] [CrossRef]
- Chae, S.; Xu, Y.; Yi, R.; Lim, H.-S.; Velickovic, D.; Li, X.; Li, Q.; Wang, C.; Zhang, J.-G. A Micrometer-Sized Silicon/Carbon Composite Anode Synthesized by Impregnation of Petroleum Pitch in Nanoporous Silicon. Adv. Mater. 2021, 33, 2103095. [Google Scholar] [CrossRef]
- Collins, G.A.; Kilian, S.; Geaney, H.; Ryan, K.M. A Nanowire Nest Structure Comprising Copper Silicide and Silicon Nanowires for Lithium-Ion Battery Anodes with High Areal Loading. Small 2021, 17, 2102333. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Shin, M.; Kim, J.; Choi, J.W.; Lee, H. Designing Adaptive Binders for Microenvironment Settings of Silicon Anode Particles. Adv. Mater. 2021, 33, 2007460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, Y.; Chen, Y.; Huang, J.; Zhang, T.; Zeng, H.; Wang, C.; Liu, G.; Deng, Y. A Quadruple-Hydrogen-Bonded Supramolecular Binder for High-Performance Silicon Anodes in Lithium-Ion Batteries. Small 2018, 14, 1801189. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.-W.; Choi, J.W.; Coskun, A. Prospect for Supramolecular Chemistry in High-Energy-Density Rechargeable Batteries. Joule 2019, 3, 662–682. [Google Scholar] [CrossRef]
- Boniface, M.; Quazuguel, L.; Danet, J.; Guyomard, D.; Moreau, P.; Bayle-Guillemaud, P. Nanoscale Chemical Evolution of Silicon Negative Electrodes Characterized by Low-Loss STEM-EELS. Nano Lett. 2016, 16, 7381–7388. [Google Scholar] [CrossRef]
- Benning, S.; Chen, C.; Eichel, R.A.; Notten, P.H.L.; Hausen, F. Direct Observation of SEI Formation and Lithiation in Thin-Film Silicon Electrodes via in Situ Electrochemical Atomic Force Microscopy. ACS Appl. Energy Mater. 2019, 2, 6761–6767. [Google Scholar] [CrossRef]
- Guo, K.; Kumar, R.; Xiao, X.; Sheldon, B.W.; Gao, H. Failure Progression in the Solid Electrolyte Interphase (SEI) on Silicon Electrodes. Nano Energy 2020, 68, 104257. [Google Scholar] [CrossRef]
- Dupré, N.; Moreau, P.; de Vito, E.; Quazuguel, L.; Boniface, M.; Bordes, A.; Rudisch, C.; Bayle-Guillemaud, P.; Guyomard, D. Multiprobe Study of the Solid Electrolyte Interphase on Silicon-Based Electrodes in Full-Cell Configuration. Chem. Mater. 2016, 28, 2557–2572. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno, C.; Mannino, G.; D’Alessio, U.; Monforte, F.; Condorelli, G.G.; Spinella, C.; La Magna, A.; Brutti, S. On the Redox Activity of the Solid Electrolyte Interphase in the Reduction/Oxidation of Silicon Nanoparticles in Secondary Lithium Batteries. Energy Technol. 2021, 10, 2100791. [Google Scholar] [CrossRef]
- Jerliu, B.; Hüger, E.; Dörrer, L.; Seidlhofer, B.K.; Steitz, R.; Oberst, V.; Geckle, U.; Bruns, M.; Schmidt, H. Volume Expansion during Lithiation of Amorphous Silicon Thin Film Electrodes Studied by In-Operando Neutron Reflectometry. J. Phys. Chem. C 2014, 118, 9395–9399. [Google Scholar] [CrossRef]
- Veryovkin, I.V.; Tripa, C.E.; Zinovev, A.V.; Baryshev, S.V.; Li, Y.; Abraham, D.P. TOF SIMS Characterization of SEI Layer on Battery Electrodes. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2014, 332, 368–372. [Google Scholar] [CrossRef]
- Browning, K.L.; Baggetto, L.; Unocic, R.R.; Dudney, N.J.; Veith, G.M. Gas Evolution from Cathode Materials: A Pathway to Solvent Decomposition Concomitant to SEI Formation. J. Power Sources 2013, 239, 341–346. [Google Scholar] [CrossRef]
- Lu, P.; Li, C.; Schneider, E.W.; Harris, S.J. Chemistry, Impedance, and Morphology Evolution in SEI Films during Formation in Lithium Ion Batteries. J. Phys. Chem. C 2013, 118, 896–903. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novák, P. A Review of the Features and Analyses of the Solid Electrolyte Interphase in Li-Ion Batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Park, S.; Jeong, S.Y.; Lee, T.K.; Park, M.W.; Lim, H.Y.; Sung, J.; Cho, J.; Kwak, S.K.; Hong, S.Y.; Choi, N.-S. Replacing Conventional Battery Electrolyte Additives with Dioxolone Derivatives for High-Energy-Density Lithium-Ion Batteries. Nat. Commun. 2021, 12, 838. [Google Scholar] [CrossRef]
- Han, B.; Zhang, Y.; Liao, C.; Trask, S.E.; Li, X.; Uppuluri, R.; Vaughey, J.T.; Key, B.; Dogan, F. Probing the Reactivity of the Active Material of a Li-Ion Silicon Anode with Common Battery Solvents. ACS Appl. Mater. Interfaces 2021, 13, 28017–28026. [Google Scholar] [CrossRef]
- Arano, K.; Begic, S.; Chen, F.; Rakov, D.; Mazouzi, D.; Gautier, N.; Kerr, R.; Lestriez, B.; Le Bideau, J.; Howlett, P.C.; et al. Tuning the Formation and Structure of the Silicon Electrode/Ionic Liquid Electrolyte Interphase in Superconcentrated Ionic Liquids. ACS Appl. Mater. Interfaces 2021, 13, 28281–28294. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Miao, M.; Li, H.; Cao, Y.; Yang, H.; Ai, X. In Situ-Formed Artificial Solid Electrolyte Interphase for Boosting the Cycle Stability of Si-Based Anodes for Li-Ion Batteries. ACS Appl. Mater. Interfaces 2021, 13, 22505–22513. [Google Scholar] [CrossRef] [PubMed]
- Roland, A.; Delarre, B.; Ledeuil, J.B.; Louvain, N.; Martinez, H.; Monconduit, L. Silicon-Based Electrodes Formulation in Buffered Solution for Enhanced Electrode-Electrolyte Interfaces. J. Power Sources 2021, 489, 229465. [Google Scholar] [CrossRef]
- Jaumann, T.; Balach, J.; Klose, M.; Oswald, S.; Eckert, J.; Giebeler, L. Role of 1,3-Dioxolane and LiNO3 Addition on the Long Term Stability of Nanostructured Silicon/Carbon Anodes for Rechargeable Lithium Batteries. J. Electrochem. Soc. 2016, 163, A557–A564. [Google Scholar] [CrossRef]
- Szczech, J.R.; Jin, S. Nanostructured Silicon for High Capacity Lithium Battery Anodes. Energy Environ. Sci. 2011, 4, 56–72. [Google Scholar] [CrossRef]
- Gao, H.; Xiao, L.; Plümel, I.; Xu, G.L.; Ren, Y.; Zuo, X.; Liu, Y.; Schulz, C.; Wiggers, H.; Amine, K.; et al. Parasitic Reactions in Nanosized Silicon Anodes for Lithium-Ion Batteries. Nano Lett. 2017, 17, 1512–1519. [Google Scholar] [CrossRef]
- Wang, J.; Xu, T.; Huang, X.; Li, H.; Ma, T. Recent Progress of Silicon Composites as Anode Materials for Secondary Batteries. RSC Adv. 2016, 6, 87778–87790. [Google Scholar] [CrossRef]
- Pan, K.; Zou, F.; Canova, M.; Zhu, Y.; Kim, J.H. Comprehensive Electrochemical Impedance Spectroscopy Study of Si-Based Anodes Using Distribution of Relaxation Times Analysis. J. Power Sources 2020, 479, 229083. [Google Scholar] [CrossRef]
- Ezzedine, M.; Zamfir, M.-R.; Jardali, F.; Leveau, L.; Caristan, E.; Ersen, O.; Cojocaru, C.-S.; Florea, I. Insight into the Formation and Stability of Solid Electrolyte Interphase for Nanostructured Silicon-Based Anode Electrodes Used in Li-Ion Batteries. ACS Appl. Mater. Interfaces 2021, 13, 24734–24746. [Google Scholar] [CrossRef]
- Nangir, M.; Massoudi, A.; Tayebifard, S.A. Investigation of the Lithium-Ion Depletion in the Silicon-Silicon Carbide Anode/Electrolyte Interface in Lithium-Ion Battery via Electrochemical Impedance Spectroscopy. J. Electroanal. Chem. 2020, 873, 114385. [Google Scholar] [CrossRef]
- Hasa, I.; Haregewoin, A.M.; Zhang, L.; Tsai, W.-Y.; Guo, J.; Veith, G.M.; Ross, P.N.; Kostecki, R. Electrochemical Reactivity and Passivation of Silicon Thin-Film Electrodes in Organic Carbonate Electrolytes. ACS Appl. Mater. Interfaces 2020, 12, 40879–40890. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, S.; Amiinu, I.S.; Storan, D.; Kapuria, N.; Geaney, H.; Kennedy, T.; Ryan, K.M. Dense Silicon Nanowire Networks Grown on a Stainless-Steel Fiber Cloth: A Flexible and Robust Anode for Lithium-Ion Batteries. Adv. Mater. 2021, 33, 2105917. [Google Scholar] [CrossRef]
- Flynn, G.; Stokes, K.; Ryan, K.M. Low Temperature Solution Synthesis of Silicon, Germanium and Si–Ge Axial Heterostructures in Nanorod and Nanowire Form. Chem. Commun. 2018, 54, 5728–5731. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.; Brandon, M.; Laffir, F.; Ryan, K.M. Understanding the Influence of Electrolyte Additives on the Electrochemical Performance and Morphology Evolution of Silicon Nanowire Based Lithium-Ion Battery Anodes. J. Power Sources 2017, 359, 601–610. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–41. [Google Scholar]
- Zhang, X.; Wang, D.; Qiu, X.; Ma, Y.; Kong, D.; Müllen, K.; Li, X.; Zhi, L. Stable High-Capacity and High-Rate Silicon-Based Lithium Battery Anodes upon Two-Dimensional Covalent Encapsulation. Nat. Commun. 2020, 11, 3826. [Google Scholar] [CrossRef]
- Fu, C.; Song, C.; Liu, L.; Zhao, W.; Xie, X. High Reversible Silicon/Graphene Nanocomposite Anode for Lithium-Ion Batteries. Int. J. Electrochem. Sci. 2016, 11, 154–164. [Google Scholar]
- Kim, G.-T.; Kennedy, T.; Brandon, M.; Geaney, H.; Ryan, K.M.; Passerini, S.; Appetecchi, G.B. Behavior of Germanium and Silicon Nanowire Anodes with Ionic Liquid Electrolytes. ACS Nano 2017, 11, 5933–5943. [Google Scholar] [CrossRef] [PubMed]
- Geaney, H.; Mullane, E.; Ryan, K.M. Solution phase synthesis of silicon and germanium nanowires. J. Mater. Chem. C 2013, 1, 4996–5007. [Google Scholar] [CrossRef]
- Smit, C.; van Swaaij, R.A.C.M.M.; Donker, H.; Petit, A.M.H.N.; Kessels, W.M.M.; van de Sanden, M.C.M. Determining the Material Structure of Microcrystalline Silicon from Raman Spectra. J. Appl. Phys. 2003, 94, 3582. [Google Scholar] [CrossRef]
- Li, B.; Yu, D.; Zhang, S. Raman Spectral Study of Silicon Nanowires. Phys. Rev. B 1999, 59, 1645. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, G.; Liu, Y.; Pan, S.; Zhang, H.; Yu, D.; Zhang, Z. Raman Spectral Study of Silicon Nanowires: High-Order Scattering and Phonon Confinement Effects. Phys. Rev. B 2000, 61, 16827. [Google Scholar] [CrossRef]
- Merlen, A.; Buijnsters, J.G.; Pardanaud, C. Coatings A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: From Graphene to Amorphous Carbons. Coatings 2017, 7, 153. [Google Scholar] [CrossRef]
- Chu, P.K.; Li, L. Characterization of Amorphous and Nanocrystalline Carbon Films. Mater. Chem. Phys. 2006, 96, 253–277. [Google Scholar] [CrossRef]
- Cabo-Fernandez, L.; Mueller, F.; Passerini, S.; Hardwick, L.J. In Situ Raman Spectroscopy of Carbon-Coated ZnFe2O4 Anode Material in Li-Ion Batteries—Investigation of SEI Growth. Chem. Commun. 2016, 52, 3970–3973. [Google Scholar] [CrossRef] [PubMed]
- Kukushkin, S.A.; Osipov, A.V. Separation of III-N/SiC Epitaxial Heterostructure from a Si Substrate and Their Transfer to Other Substrate Types. Semiconductors 2017, 51, 396–401. [Google Scholar] [CrossRef]
- Seidlhofer, B.K.; Jerliu, B.; Trapp, M.; Hüger, E.; Risse, S.; Cubitt, R.; Schmidt, H.; Steitz, R.; Ballauff, M. Lithiation of Crystalline Silicon As Analyzed by Operando Neutron Reflectivity. ACS Nano 2016, 10, 7458–7466. [Google Scholar] [CrossRef]
- Graf, M.; Berg, C.; Bernhard, R.; Haufe, S.; Pfeiffer, J.; Gasteiger, H.A. Effect and Progress of the Amorphization Process for Microscale Silicon Particles under Partial Lithiation as Active Material in Lithium-Ion Batteries. J. Electrochem. Soc. 2022, 169, 020536. [Google Scholar] [CrossRef]
- Obrovac, M.N.; Christensen, L. Structural Changes in Silicon Anodes during Lithium Insertion/Extraction. Electrochem. Solid-State Lett. 2004, 7, A93. [Google Scholar] [CrossRef]
- Tang, M.; Newman, J. Why is the Solid-Electrolyte-Interphase Selective? Through-Film Ferrocenium Reduction on Highly Oriented Pyrolytic Graphite. J. Electrochem. Soc. 2012, 159, A1922. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Winter, M. Advances in Battery Technology: Rechargeable Magnesium Batteries and Novel Negative-Electrode Materials for Lithium Ion Batteries. ChemPhysChem 2002, 3, 155–159. [Google Scholar] [CrossRef]
- Attia, P.M.; Das, S.; Harris, S.; Bazant, M.Z.; Chueh, W.C. Electrochemical Kinetics of SEI Growth on Carbon Black: Part I. Experiments. J. Electrochem. Soc. 2019, 166, E97. [Google Scholar] [CrossRef]
- Borodin, O.; Ren, X.; Vatamanu, J.; von Wald Cresce, A.; Knap, J.; Xu, K. Modeling Insight into Battery Electrolyte Electrochemical Stability and Interfacial Structure. Acc. Chem. Res. 2017, 50, 2886–2894. [Google Scholar] [CrossRef]
- Huang, W.; Wang, J.; Braun, M.R.; Zhang, Z.; Li, Y.; Boyle, D.T.; McIntyre, P.C.; Cui, Y. Dynamic Structure and Chemistry of the Silicon Solid-Electrolyte Interphase Visualized by Cryogenic Electron Microscopy. Matter 2019, 1, 1232–1245. [Google Scholar] [CrossRef]
- Zhuo, Z.; Lu, P.; Delacourt, C.; Qiao, R.; Xu, K.; Pan, F.; Harris, S.J.; Yang, W. Breathing and oscillating growth of solid-electrolyte-interphase upon electrochemical cycling. Chem. Commun. 2018, 54, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, L.; Bi, X.; Tian, L.; Liu, J.; Li, M.; Chen, Z.; Lu, J.; Amine, K.; Xu, K.; et al. In situ quantification of interphasial chemistry in Li-ion battery. Nat. Nanotech 2019, 14, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ahoulou, S.; Perret, E.; Nedelec, J.M. Functionalization and Characterization of Silicon Nanowires for Sensing Applications: A Review. Nanomaterials 2021, 11, 999. [Google Scholar] [CrossRef]
- Profatilova, I.A.; Stock, C.; Schmitz, A.; Passerini, S.; Winter, M. Enhanced Thermal Stability of a Lithiated Nano-Silicon Electrode by Fluoroethylene Carbonate and Vinylene Carbonate. J. Power Sources 2013, 222, 140–149. [Google Scholar] [CrossRef]
- Shi, F.; Ross, P.N.; Somorjai, G.A.; Komvopoulos, K. The Chemistry of Electrolyte Reduction on Silicon Electrodes Revealed by in Situ ATR-FTIR Spectroscopy. J. Phys. Chem. C 2017, 121, 14476–14483. [Google Scholar] [CrossRef]
- Kleinová, A.; Huran, J.; Sasinková, V.; Perný, M.; Šály, V.; Packa, J. FTIR Spectroscopy of Silicon Carbide Thin Films Prepared by PECVD Technology for Solar Cell Application. In Proceedings of the Reliability of Photovoltaic Cells, Modules, Components, and Systems VIII, San Diego, CA, USA, 9–13 August 2015; Volume 9563, pp. 166–173. [Google Scholar] [CrossRef]
- Key, B.; Bhattacharyya, R.; Morcrette, M.; Seznéc, V.; Tarascon, J.M.; Grey, C.P. Real-Time NMR Investigations of Structural Changes in Silicon Electrodes for Lithium-Ion Batteries. J. Am. Chem. Soc. 2009, 131, 9239–9249. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pristine Si NWs 49 | |||
|---|---|---|---|
| Post mortem samples | After 5 cycles | After 10 cycles | After 50 cycles |
| BE | 57 | 80 | 94 |
| FEC-added electrolyte | 52 | 71 | 89 |
| VC-added electrolyte | 66 | 90 | 94 |
| Electrolyte | Lithiation Reactions (Reductions, Discharges) | De-Lithiation Reactions (Oxidations, Charges) | ||||
|---|---|---|---|---|---|---|
| Qa-Si,red | Qc-Si,red | QSEI,red | Qa-Si,red,net | QLi-trapping | QSEI,ox | |
| BE | 82% | 3% | 16% | 83% | 1% | 6% |
| FEC-added electrolyte | 67% | 1% | 32% | 68% | 0% | 24% |
| VC-added electrolyte | 73% | 4% | 22% | 78% | 0% | 14% |
| Sample | Amorphization | Carbonate Content | LiPF6 Decomposition Residues Content | |
|---|---|---|---|---|
| uncycled | low | absent | absent | |
| post mortem | BE | medium | high | high |
| FEC-added electrolyte | low | medium | medium | |
| VC-added electrolyte | high | medium | low | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarra, A.; Brutti, S.; Palumbo, O.; Capitani, F.; Borondics, F.; Appetecchi, G.B.; Carboni, N.; Abdul Ahad, S.; Geaney, H.; Ryan, K.; et al. Solid–Electrolyte Interface Formation on Si Nanowires in Li-Ion Batteries: The Impact of Electrolyte Additives. Batteries 2023, 9, 148. https://doi.org/10.3390/batteries9030148
Sarra A, Brutti S, Palumbo O, Capitani F, Borondics F, Appetecchi GB, Carboni N, Abdul Ahad S, Geaney H, Ryan K, et al. Solid–Electrolyte Interface Formation on Si Nanowires in Li-Ion Batteries: The Impact of Electrolyte Additives. Batteries. 2023; 9(3):148. https://doi.org/10.3390/batteries9030148
Chicago/Turabian StyleSarra, Angelo, Sergio Brutti, Oriele Palumbo, Francesco Capitani, Ferenc Borondics, Giovanni Battista Appetecchi, Nicholas Carboni, Syed Abdul Ahad, Hugh Geaney, Kevin Ryan, and et al. 2023. "Solid–Electrolyte Interface Formation on Si Nanowires in Li-Ion Batteries: The Impact of Electrolyte Additives" Batteries 9, no. 3: 148. https://doi.org/10.3390/batteries9030148
APA StyleSarra, A., Brutti, S., Palumbo, O., Capitani, F., Borondics, F., Appetecchi, G. B., Carboni, N., Abdul Ahad, S., Geaney, H., Ryan, K., & Paolone, A. (2023). Solid–Electrolyte Interface Formation on Si Nanowires in Li-Ion Batteries: The Impact of Electrolyte Additives. Batteries, 9(3), 148. https://doi.org/10.3390/batteries9030148